 |
PDBsum entry 2v4l

|
|
|
|
|
 |
Contents |
 |
|
|
|
|
|
|
|
|
|
|
|
|
* Residue conservation analysis
|
|
|
|
|
|
PDB id:
|
|
|
|
| Name: |
|
Transferase
|
|
|
Title:
|
|
Complex of human phosphoinositide 3-kinase catalytic subunit gamma (p110 gamma) with pik-284
|
|
Structure:
|
|
Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit gamma isoform. Chain: a. Fragment: catalytic subunit, residues 144-1102. Synonym: pi3-kinase p110 subunit gamma, ptdins-3-kinase subunit p110, pi3kgamma, p120-pi3k, pi3k, phosphoinositide 3-kinase gamma. Engineered: yes
|
|
Source:
|
|
Homo sapiens. Human. Organism_taxid: 9606. Expressed in: spodoptera frugiperda. Expression_system_taxid: 7108.
|
|
Resolution:
|
|
|
2.50Å
|
R-factor:
|
0.250
|
R-free:
|
0.296
|
|
|
Authors:
|
|
B.Apsel,B.Gonzalez,J.A.Blair,T.M.Nazif,M.E.Feldman,R.L.Williams, K.M.Shokat,Z.A.Knight
|
Key ref:

|
|
B.Apsel
et al.
(2008).
Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases.
Nat Chem Biol,
4,
691-699.
PubMed id:
DOI:

|
|
|
Date:
|
|
|
25-Sep-08
|
Release date:
|
14-Oct-08
|
|
|
|
|
|
|
PROCHECK
|

|
|
|
|
|
Headers
|
|
|
|
References
|
|
|
|
|
|

|
|
P48736
(PK3CG_HUMAN) -
Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit gamma isoform from Homo sapiens
|
|
|
|
Seq:
Struc:
|
|
|
|
1102 a.a.
845 a.a.*
|
|

|

|
|
|
|
|
|
|
|
|
|
|
|
Key: |
 |
PfamA domain |
|
|
 |
Secondary structure |
|
 |
CATH domain |
|
|
*
PDB and UniProt seqs differ
at 1 residue position (black
cross)
|
|
|
|
|
|
|
|
|
|
|
|
|
Enzyme class 2:
|
|
E.C.2.7.11.1
- non-specific serine/threonine protein kinase.
|
|
|
|
|
|
|
Reaction:
|
|
|
1.
|
L-seryl-[protein] + ATP = O-phospho-L-seryl-[protein] + ADP + H+
|
|
2.
|
L-threonyl-[protein] + ATP = O-phospho-L-threonyl-[protein] + ADP + H+
|
|
|
|
|
|
|
L-seryl-[protein]
|
+
|
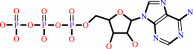 ATP
ATP
|
=
|
O-phospho-L-seryl-[protein]
|
+
|
 ADP
ADP
|
+
|
H(+)
|
|
|
|
|
|
|
L-threonyl-[protein]
|
+
|
ATP
|
=
|
O-phospho-L-threonyl-[protein]
|
+
|
ADP
|
+
|
H(+)
|
|
|
|
|
|
|
|
|
|
Enzyme class 3:
|
|
E.C.2.7.1.137
- phosphatidylinositol 3-kinase.
|
|
|
|
|
|
|
Pathway:
|
|
|
|
|
|
|
|
Reaction:
|
|
a 1,2-diacyl-sn-glycero-3-phospho-(1D-myo-inositol) + ATP = a 1,2-diacyl- sn-glycero-3-phospho-(1D-myo-inositol-3-phosphate) + ADP + H+
|
|
|
|
|
|
1,2-diacyl-sn-glycero-3-phospho-(1D-myo-inositol)
|
+
|
ATP
|
=
|
1,2-diacyl- sn-glycero-3-phospho-(1D-myo-inositol-3-phosphate)
|
+
|
ADP
|
+
|
H(+)
|
|
|
|
|
|
|
|
|
|
Enzyme class 4:
|
|
E.C.2.7.1.153
- phosphatidylinositol-4,5-bisphosphate 3-kinase.
|
|
|
|
|
|
|
Pathway:
|
|
|
|
|
|
|
|
Reaction:
|
|
a 1,2-diacyl-sn-glycero-3-phospho-(1D-myo-inositol-4,5-bisphosphate) + ATP = a 1,2-diacyl-sn-glycero-3-phospho-(1D-myo-inositol-3,4,5- trisphosphate) + ADP + H+
|
|
|
|
|
|
1,2-diacyl-sn-glycero-3-phospho-(1D-myo-inositol-4,5-bisphosphate)
|
+
|
ATP
|
=
|
1,2-diacyl-sn-glycero-3-phospho-(1D-myo-inositol-3,4,5- trisphosphate)
|
+
|
ADP
|
+
|
H(+)
|
|
|
|
|
|
|
|
|
|
Enzyme class 5:
|
|
E.C.2.7.1.154
- phosphatidylinositol-4-phosphate 3-kinase.
|
|
|
|
|
|
|
Pathway:
|
|
|
|
|
|
|
|
Reaction:
|
|
a 1,2-diacyl-sn-glycero-3-phospho-(1D-myo-inositol 4-phosphate) + ATP = a 1,2-diacyl-sn-glycero-3-phospho-(1D-myo-inositol-3,4-bisphosphate) + ADP + H+
|
|
|
|
|
|
1,2-diacyl-sn-glycero-3-phospho-(1D-myo-inositol 4-phosphate)
|
+
|
ATP
|
=
|
1,2-diacyl-sn-glycero-3-phospho-(1D-myo-inositol-3,4-bisphosphate)
|
+
|
ADP
|
+
|
H(+)
|
|
|
|
|
|
|
|
|
|
|
|
|
Note, where more than one E.C. class is given (as above), each may
correspond to a different protein domain or, in the case of polyprotein
precursors, to a different mature protein.
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
DOI no:
|
Nat Chem Biol
4:691-699
(2008)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases.
|
|
B.Apsel,
J.A.Blair,
B.Gonzalez,
T.M.Nazif,
M.E.Feldman,
B.Aizenstein,
R.Hoffman,
R.L.Williams,
K.M.Shokat,
Z.A.Knight.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
The clinical success of multitargeted kinase inhibitors has stimulated efforts
to identify promiscuous drugs with optimal selectivity profiles. It remains
unclear to what extent such drugs can be rationally designed, particularly for
combinations of targets that are structurally divergent. Here we report the
systematic discovery of molecules that potently inhibit both tyrosine kinases
and phosphatidylinositol-3-OH kinases, two protein families that are among the
most intensely pursued cancer drug targets. Through iterative chemical
synthesis, X-ray crystallography and kinome-level biochemical profiling, we
identified compounds that inhibit a spectrum of new target combinations in these
two families. Crystal structures revealed that the dual selectivity of these
molecules is controlled by a hydrophobic pocket conserved in both enzyme classes
and accessible through a rotatable bond in the drug skeleton. We show that one
compound, PP121, blocks the proliferation of tumor cells by direct inhibition of
oncogenic tyrosine kinases and phosphatidylinositol-3-OH kinases. These
molecules demonstrate the feasibility of accessing a chemical space that
intersects two families of oncogenes.
|
|
|
|
|
|
| |
Selected figure(s)
|
|
|
|
| |
|
|

|
|

|
|
Figure 2.
(a) Experimental strategy for the discovery of dual
inhibitors, and IC[50] values (  M)
for 8 molecules tested against 14 tyrosine kinases and PI(3)Ks
(10 M
ATP). IC[50] values less than 0.1 M
are shaded red. Pyrazolopyrimidine N4 and N5, which make
hydrogen bonds to the kinase, are labeled. (b) Percentage
inhibition of 84 tyrosine kinases (right) and 135
serine-threonine kinases (left) by 7 inhibitors from this study
(right columns) and 5 reference compounds (left columns). PP
inhibitors were tested at 1 M
drug and, typically, 10 M
ATP. Data from the Invitrogen SelectScreen assay. (c) Principal
component analysis of the target selectivity of 172
pyrazolopyrimidine inhibitors and 8 reference compounds. Key
compounds are labeled. M)
for 8 molecules tested against 14 tyrosine kinases and PI(3)Ks
(10 M
ATP). IC[50] values less than 0.1 M
are shaded red. Pyrazolopyrimidine N4 and N5, which make
hydrogen bonds to the kinase, are labeled. (b) Percentage
inhibition of 84 tyrosine kinases (right) and 135
serine-threonine kinases (left) by 7 inhibitors from this study
(right columns) and 5 reference compounds (left columns). PP
inhibitors were tested at 1 M
drug and, typically, 10 M
ATP. Data from the Invitrogen SelectScreen assay. (c) Principal
component analysis of the target selectivity of 172
pyrazolopyrimidine inhibitors and 8 reference compounds. Key
compounds are labeled.
|
|
Figure 4.
(a) Correlation between IC[50] values for inhibitors against
Src (x axis) and either Hck or the gatekeeper mutant Src T338I
(y axis). (b) Binding orientation of S1 relative to ATP in c-Src
(top) and p110  (bottom).
(c) Overlay of cocrystal structures of inhibitors bound to c-Src
(protein colored red, drugs orange: S1, PP102, PP121 and PP494)
and p110 (protein
blue, compounds gray: S1 and S2). The gatekeeper residues Thr338
(c-Src) and Ile879 (p110 )
are highlighted. (d) Top, the catalytic lysine (Lys295) makes a
hydrogen bond to Glu310 in active c-Src. Center, helix C and
Glu310 are disordered in c-Src structures containing PP102.
Bottom, PP121 makes a hydrogen bond to Glu310 and orders helix C
when bound to c-Src. (bottom).
(c) Overlay of cocrystal structures of inhibitors bound to c-Src
(protein colored red, drugs orange: S1, PP102, PP121 and PP494)
and p110 (protein
blue, compounds gray: S1 and S2). The gatekeeper residues Thr338
(c-Src) and Ile879 (p110 )
are highlighted. (d) Top, the catalytic lysine (Lys295) makes a
hydrogen bond to Glu310 in active c-Src. Center, helix C and
Glu310 are disordered in c-Src structures containing PP102.
Bottom, PP121 makes a hydrogen bond to Glu310 and orders helix C
when bound to c-Src.
|
|
|
|
|
|
| |
The above figures are
reprinted
by permission from Macmillan Publishers Ltd:
Nat Chem Biol
(2008,
4,
691-699)
copyright 2008.
|
|
| |
Figures were
selected
by an automated process.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|
Literature references that cite this PDB file's key reference
|
|
|
| |
PubMed id
|
|
Reference
|
|
|
|
|
|
A.Dormond-Meuwly,
D.Roulin,
M.Dufour,
M.Benoit,
N.Demartines,
and
O.Dormond
(2011).
The inhibition of MAPK potentiates the anti-angiogenic efficacy of mTOR inhibitors.
|
| |
Biochem Biophys Res Commun,
407,
714-719.
|
|
|
|
|
|
|
C.Garcia-Echeverria
(2011).
Blocking the mTOR pathway: a drug discovery perspective.
|
| |
Biochem Soc Trans,
39,
451-455.
|
|
|
|
|
|
|
H.Jo,
P.K.Lo,
Y.Li,
F.Loison,
S.Green,
J.Wang,
L.E.Silberstein,
K.Ye,
H.Chen,
and
H.R.Luo
(2011).
Deactivation of Akt by a small molecule inhibitor targeting pleckstrin homology domain and facilitating Akt ubiquitination.
|
| |
Proc Natl Acad Sci U S A,
108,
6486-6491.
|
|
|
|
|
|
|
H.Yabuuchi,
S.Niijima,
H.Takematsu,
T.Ida,
T.Hirokawa,
T.Hara,
T.Ogawa,
Y.Minowa,
G.Tsujimoto,
and
Y.Okuno
(2011).
Analysis of multiple compound-protein interactions reveals novel bioactive molecules.
|
| |
Mol Syst Biol,
7,
472.
|
|
|
|
|
|
|
I.Takigawa,
K.Tsuda,
and
H.Mamitsuka
(2011).
Mining significant substructure pairs for interpreting polypharmacology in drug-target network.
|
| |
PLoS One,
6,
e16999.
|
|
|
|
|
|
|
J.R.Brown,
and
K.R.Auger
(2011).
Phylogenomics of phosphoinositide lipid kinases: perspectives on the evolution of second messenger signaling and drug discovery.
|
| |
BMC Evol Biol,
11,
4.
|
|
|
|
|
|
|
S.Schenone,
O.Bruno,
M.Radi,
and
M.Botta
(2011).
New insights into small-molecule inhibitors of Bcr-Abl.
|
| |
Med Res Rev,
31,
1.
|
|
|
|
|
|
|
U.Kruse,
C.P.Pallasch,
M.Bantscheff,
D.Eberhard,
L.Frenzel,
S.Ghidelli,
S.K.Maier,
T.Werner,
C.M.Wendtner,
and
G.Drewes
(2011).
Chemoproteomics-based kinome profiling and target deconvolution of clinical multi-kinase inhibitors in primary chronic lymphocytic leukemia cells.
|
| |
Leukemia,
25,
89.
|
|
|
|
|
|
|
Y.J.Zhang,
Y.Duan,
and
X.F.Zheng
(2011).
Targeting the mTOR kinase domain: the second generation of mTOR inhibitors.
|
| |
Drug Discov Today,
16,
325-331.
|
|
|
|
|
|
|
A.Berndt,
S.Miller,
O.Williams,
D.D.Le,
B.T.Houseman,
J.I.Pacold,
F.Gorrec,
W.C.Hon,
Y.Liu,
C.Rommel,
P.Gaillard,
T.Rückle,
M.K.Schwarz,
K.M.Shokat,
J.P.Shaw,
and
R.L.Williams
(2010).
The p110 delta structure: mechanisms for selectivity and potency of new PI(3)K inhibitors.
|
| |
Nat Chem Biol,
6,
117-124.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
A.Pujol,
R.Mosca,
J.Farrés,
and
P.Aloy
(2010).
Unveiling the role of network and systems biology in drug discovery.
|
| |
Trends Pharmacol Sci,
31,
115-123.
|
|
|
|
|
|
|
M.R.Janes,
J.J.Limon,
L.So,
J.Chen,
R.J.Lim,
M.A.Chavez,
C.Vu,
M.B.Lilly,
S.Mallya,
S.T.Ong,
M.Konopleva,
M.B.Martin,
P.Ren,
Y.Liu,
C.Rommel,
and
D.A.Fruman
(2010).
Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor.
|
| |
Nat Med,
16,
205-213.
|
|
|
|
|
|
|
S.B.Gabelli,
D.Mandelker,
O.Schmidt-Kittler,
B.Vogelstein,
and
L.M.Amzel
(2010).
Somatic mutations in PI3Kalpha: structural basis for enzyme activation and drug design.
|
| |
Biochim Biophys Acta,
1804,
533-540.
|
|
|
|
|
|
|
V.R.Konda,
A.Desai,
G.Darland,
J.S.Bland,
and
M.L.Tripp
(2010).
META060 inhibits osteoclastogenesis and matrix metalloproteinases in vitro and reduces bone and cartilage degradation in a mouse model of rheumatoid arthritis.
|
| |
Arthritis Rheum,
62,
1683-1692.
|
|
|
|
|
|
|
Z.A.Knight,
H.Lin,
and
K.M.Shokat
(2010).
Targeting the cancer kinome through polypharmacology.
|
| |
Nat Rev Cancer,
10,
130-137.
|
|
|
|
|
|
|
I.D.Fraser,
and
R.N.Germain
(2009).
Navigating the network: signaling cross-talk in hematopoietic cells.
|
| |
Nat Immunol,
10,
327-331.
|
|
|
|
|
|
|
J.Lehár,
A.S.Krueger,
W.Avery,
A.M.Heilbut,
L.M.Johansen,
E.R.Price,
R.J.Rickles,
G.F.Short,
J.E.Staunton,
X.Jin,
M.S.Lee,
G.R.Zimmermann,
and
A.A.Borisy
(2009).
Synergistic drug combinations tend to improve therapeutically relevant selectivity.
|
| |
Nat Biotechnol,
27,
659-666.
|
|
|
|
|
|
|
K.Ghoreschi,
A.Laurence,
and
J.J.O'Shea
(2009).
Selectivity and therapeutic inhibition of kinases: to be or not to be?
|
| |
Nat Immunol,
10,
356-360.
|
|
|
|
|
|
|
L.A.Smyth,
and
I.Collins
(2009).
Measuring and interpreting the selectivity of protein kinase inhibitors.
|
| |
J Chem Biol,
2,
131-151.
|
|
|
|
|
|
|
M.E.Feldman,
B.Apsel,
A.Uotila,
R.Loewith,
Z.A.Knight,
D.Ruggero,
and
K.M.Shokat
(2009).
Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2.
|
| |
PLoS Biol,
7,
e38.
|
|
|
|
|
|
|
M.T.Grande,
and
J.M.López-Novoa
(2009).
Fibroblast activation and myofibroblast generation in obstructive nephropathy.
|
| |
Nat Rev Nephrol,
5,
319-328.
|
|
|
|
|
|
|
N.P.Tatonetti,
T.Liu,
and
R.B.Altman
(2009).
Predicting drug side-effects by chemical systems biology.
|
| |
Genome Biol,
10,
238.
|
|
|
|
|
|
|
P.Liu,
H.Cheng,
T.M.Roberts,
and
J.J.Zhao
(2009).
Targeting the phosphoinositide 3-kinase pathway in cancer.
|
| |
Nat Rev Drug Discov,
8,
627-644.
|
|
|
|
|
|
|
Q.Liu,
C.Thoreen,
J.Wang,
D.Sabatini,
and
N.S.Gray
(2009).
mTOR Mediated Anti-Cancer Drug Discovery.
|
| |
Drug Discov Today Ther Strateg,
6,
47-55.
|
|
|
|
|
|
|
R.L.van Montfort,
and
P.Workman
(2009).
Structure-based design of molecular cancer therapeutics.
|
| |
Trends Biotechnol,
27,
315-328.
|
|
|
|
|
|
|
T.Okuzumi,
D.Fiedler,
C.Zhang,
D.C.Gray,
B.Aizenstein,
R.Hoffman,
and
K.M.Shokat
(2009).
Inhibitor hijacking of Akt activation.
|
| |
Nat Chem Biol,
5,
484-493.
|
|
|
|
|
|
|
T.W.Sturgill,
and
M.N.Hall
(2009).
Activating mutations in TOR are in similar structures as oncogenic mutations in PI3KCalpha.
|
| |
ACS Chem Biol,
4,
999.
|
|
|
|
|
|
|
Y.Wang,
A.F.Monzingo,
S.Hu,
T.H.Schaller,
J.D.Robertus,
and
W.Fast
(2009).
Developing dual and specific inhibitors of dimethylarginine dimethylaminohydrolase-1 and nitric oxide synthase: toward a targeted polypharmacology to control nitric oxide.
|
| |
Biochemistry,
48,
8624-8635.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
B.Bilanges,
N.Torbett,
and
B.Vanhaesebroeck
(2008).
Killing two kinase families with one stone.
|
| |
Nat Chem Biol,
4,
648-649.
|
|
|
|
|
|
The most recent references are shown first.
Citation data come partly from CiteXplore and partly
from an automated harvesting procedure. Note that this is likely to be
only a partial list as not all journals are covered by
either method. However, we are continually building up the citation data
so more and more references will be included with time.
Where a reference describes a PDB structure, the PDB
codes are
shown on the right.
|
|


 Links
Links








