 |
PDBsum entry 1msm

|
|
|
|
|
|

|

|

|

|
|

|
|

|
|

|
|

|
|
|
 |

|

|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Hydrolase/hydrolase inhibitor
|
PDB id
|
|
|
|
1msm
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

|

|

|
|
|
|
|
|
|
|
 |
Contents |
 |
|
|
|
|
|
|
|
|
|
|
|
|
* Residue conservation analysis
|
|
|
|
|
|
|
|
|
|
|
Enzyme class 1:
|
|
E.C.2.7.7.-
- ?????
|
|
|
|
|
|
|
Enzyme class 2:
|
|
E.C.2.7.7.49
- RNA-directed Dna polymerase.
|
|
|
|
|
|
|
Reaction:
|
|
DNA(n) + a 2'-deoxyribonucleoside 5'-triphosphate = DNA(n+1) + diphosphate
|
|
|
|
|
|
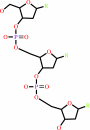 DNA(n)
DNA(n)
|
+
|
2'-deoxyribonucleoside 5'-triphosphate
|
=
|
DNA(n+1)
|
+
|
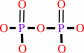 diphosphate
diphosphate
|
|
|
|
|
|
|
|
|
|
Enzyme class 3:
|
|
E.C.2.7.7.7
- DNA-directed Dna polymerase.
|
|
|
|
|
|
|
Reaction:
|
|
DNA(n) + a 2'-deoxyribonucleoside 5'-triphosphate = DNA(n+1) + diphosphate
|
|
|
|
|
|
DNA(n)
|
+
|
2'-deoxyribonucleoside 5'-triphosphate
|
=
|
DNA(n+1)
|
+
|
diphosphate
|
|
|
|
|
|
|
|
|
|
Enzyme class 4:
|
|
E.C.3.1.-.-
|
|
|
|
|
|
|
Enzyme class 5:
|
|
E.C.3.1.13.2
- exoribonuclease H.
|
|
|
|
|
|
|
Reaction:
|
|
Exonucleolytic cleavage to 5'-phosphomonoester oligonucleotides in both 5'- to 3'- and 3'- to 5'-directions.
|
|
|
|
|
|
Enzyme class 6:
|
|
E.C.3.1.26.13
- retroviral ribonuclease H.
|
|
|
|
|
|
|
Enzyme class 7:
|
|
E.C.3.4.23.16
- HIV-1 retropepsin.
|
|
|
|
|
|
|
Reaction:
|
|
Specific for a P1 residue that is hydrophobic, and P1' variable, but often Pro.
|
|
|
|
|
|
|
|
|
Note, where more than one E.C. class is given (as above), each may
correspond to a different protein domain or, in the case of polyprotein
precursors, to a different mature protein.
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
DOI no:
|
Proteins
55:594-602
(2004)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
A structural and thermodynamic escape mechanism from a drug resistant mutation of the HIV-1 protease.
|
|
S.Vega,
L.W.Kang,
A.Velazquez-Campoy,
Y.Kiso,
L.M.Amzel,
E.Freire.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
The efficacy of HIV-1 protease inhibition therapies is often compromised by the
appearance of mutations in the protease molecule that lower the binding affinity
of inhibitors while maintaining viable catalytic activity and substrate
affinity. The V82F/I84V double mutation is located within the binding site
cavity and affects all protease inhibitors in clinical use. KNI-764, a
second-generation inhibitor currently under development, maintains significant
potency against this mutation by entropically compensating for enthalpic losses,
thus minimizing the loss in binding affinity. KNI-577 differs from KNI-764 by a
single functional group critical to the inhibitor response to the protease
mutation. This single difference changes the response of the two inhibitors to
the mutation by one order of magnitude. Accordingly, a structural understanding
of the inhibitor response will provide important guidelines for the design of
inhibitors that are less susceptible to mutations conveying drug resistance. The
structures of the two compounds bound to the wild type and V82F/I84V HIV-1
protease have been determined by X-ray crystallography at 2.0 A resolution. The
presence of two asymmetric functional groups, linked by rotatable bonds to the
inhibitor scaffold, allows KNI-764 to adapt to the mutated binding site cavity
more readily than KNI-577, with a single asymmetric group. Both inhibitors lose
about 2.5 kcal/mol in binding enthalpy when facing the drug-resistant mutant
protease; however KNI-764 gains binding entropy while KNI-577 loses binding
entropy. The gain in binding entropy by KNI-764 accounts for its low
susceptibility to the drug-resistant mutation. The heat capacity change
associated with binding becomes more negative when KNI-764 binds to the mutant
protease, consistent with increased desolvation. With KNI-577, the opposite
effect is observed. Structurally, the crystallographic B factors increase for
KNI-764 when it is bound to the drug-resistant mutant. The opposite is observed
for KNI-577. Consistent with these observations, it appears that KNI-764 is able
to gain binding entropy by a two-fold mechanism: it gains solvation entropy by
burying itself deeper within the binding pocket and gains conformational entropy
by losing interaction with the protease.
|
|
|
|
|
|
| |
Selected figure(s)
|
|
|
|
| |
|
|

|
|

|
|
Figure 2.
Figure 2. The chemical structure of KNI-577 (left) and KNI-764
(right). Both inhibitors share the same allophenyl-norstatine
scaffold at the P1 position (red) and the same functional groups
at the P2 (blue) and P1  positions
(green). The only difference is at the P2 position
(magenta). positions
(green). The only difference is at the P2 position
(magenta).
|
|
Figure 4.
Figure 4. Superposition of the complexes of KNI-764 (left) and
KNI-577 (right) bound to wild-type (cyan) and double mutant
(purple) protease.
|
|
|
|
|
|
| |
The above figures are
reprinted
by permission from John Wiley & Sons, Inc.:
Proteins
(2004,
55,
594-602)
copyright 2004.
|
|
| |
Figures were
selected
by an automated process.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|
Literature references that cite this PDB file's key reference
|
|
|
| |
PubMed id
|
|
Reference
|
|
|
|
|
|
Y.Kawasaki,
E.E.Chufan,
V.Lafont,
K.Hidaka,
Y.Kiso,
L.Mario Amzel,
and
E.Freire
(2010).
How much binding affinity can be gained by filling a cavity?
|
| |
Chem Biol Drug Des,
75,
143-151.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
D.Das,
Y.Koh,
Y.Tojo,
A.K.Ghosh,
and
H.Mitsuya
(2009).
Prediction of potency of protease inhibitors using free energy simulations with polarizable quantum mechanics-based ligand charges and a hybrid water model.
|
| |
J Chem Inf Model,
49,
2851-2862.
|
|
|
|
|
|
|
D.Li,
M.S.Liu,
B.Ji,
K.Hwang,
and
Y.Huang
(2009).
Coarse-grained molecular dynamics of ligands binding into protein: The case of HIV-1 protease inhibitors.
|
| |
J Chem Phys,
130,
215102.
|
|
|
|
|
|
|
E.Freire
(2009).
A thermodynamic approach to the affinity optimization of drug candidates.
|
| |
Chem Biol Drug Des,
74,
468-472.
|
|
|
|
|
|
|
E.T.Brower,
U.M.Bacha,
Y.Kawasaki,
and
E.Freire
(2008).
Inhibition of HIV-2 protease by HIV-1 protease inhibitors in clinical use.
|
| |
Chem Biol Drug Des,
71,
298-305.
|
|
|
|
|
|
|
J.T.Nguyen,
Y.Hamada,
T.Kimura,
and
Y.Kiso
(2008).
Design of potent aspartic protease inhibitors to treat various diseases.
|
| |
Arch Pharm (Weinheim),
341,
523-535.
|
|
|
|
|
|
|
S.Muzammil,
A.A.Armstrong,
L.W.Kang,
A.Jakalian,
P.R.Bonneau,
V.Schmelmer,
L.M.Amzel,
and
E.Freire
(2007).
Unique thermodynamic response of tipranavir to human immunodeficiency virus type 1 protease drug resistance mutations.
|
| |
J Virol,
81,
5144-5154.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
V.Hornak,
and
C.Simmerling
(2007).
Targeting structural flexibility in HIV-1 protease inhibitor binding.
|
| |
Drug Discov Today,
12,
132-138.
|
|
|
|
|
|
|
V.Lafont,
A.A.Armstrong,
H.Ohtaka,
Y.Kiso,
L.Mario Amzel,
and
E.Freire
(2007).
Compensating enthalpic and entropic changes hinder binding affinity optimization.
|
| |
Chem Biol Drug Des,
69,
413-422.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
S.Avram,
C.Bologa,
and
M.L.Flonta
(2005).
Quantitative structure-activity relationship by CoMFA for cyclic urea and nonpeptide-cyclic cyanoguanidine derivatives on wild type and mutant HIV-1 protease.
|
| |
J Mol Model,
11,
105-115.
|
|
|
|
|
|
|
H.M.Abdel-Rahman,
T.Kimura,
K.Hidaka,
A.Kiso,
A.Nezami,
E.Freire,
Y.Hayashi,
and
Y.Kiso
(2004).
Design of inhibitors against HIV, HTLV-I, and Plasmodium falciparum aspartic proteases.
|
| |
Biol Chem,
385,
1035-1039.
|
|
|
|
|
|
The most recent references are shown first.
Citation data come partly from CiteXplore and partly
from an automated harvesting procedure. Note that this is likely to be
only a partial list as not all journals are covered by
either method. However, we are continually building up the citation data
so more and more references will be included with time.
Where a reference describes a PDB structure, the PDB
codes are
shown on the right.
|
|


 Links
Links
















