
Kiyosu Medaka Population
Locality

The Kiyosu site irrigation channel.
We sampled fish from two Southern Japanese locations close to Nagoya where the NIBB Medaka resource group under Kiyoshi Naruse is housed: irrigation canals in the Takashi Hongo and the Kiyosu areas near Toyohashi (Aichi Prefecture) in July 2010. Following analysis of the mitotype of individuals from the two sites, we rejected the Takashi Hongoe site in favour of Kiyosu. See the sampling page for more on locations and the sampling expedition.
The Kiyosu sampling site lies on an island formed at the exit of the Toyokawa river into Mikawa Bay, between the municipalities of Toyokawa and Toyohashi in Aichi prefecture. The site itself is an irrigation channel surrounded by rice paddies close to tidal water.
Genetics
Population Structure
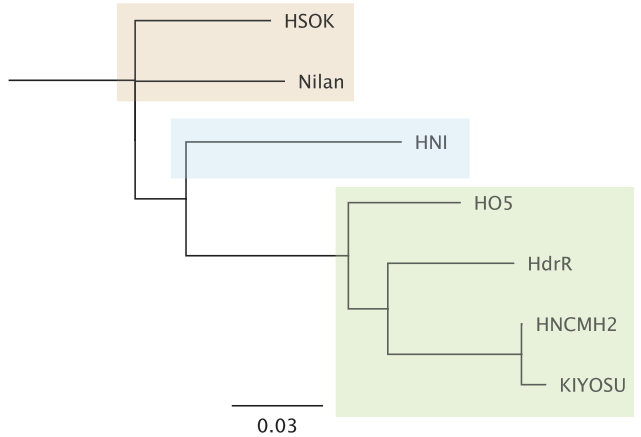
The Kiyosu population clusters with Southern inbred lines by D loop sequencing.
We sequenced the highly variable D-loop region of the mitochondrial cytochrome B gene in 105 Kiyosu individuals and detected 8 different sequence variants. This degree of D-loop sequence diversity was comparable to an earlier study where Katsumura et al. (Katsumura et al. 2009) detected 11 sequence types in 124 individuals in the most diverse and 3 types in 159 individuals in the least diverse of three wild medaka populations. Compared to the established Medaka inbred strains, the Kiyosu population clustered with the southern HdrR, HO5 and HNCMH2 strains as expected.
We analyzed microsatellite markers from 9 of the 24 medaka linkage groups and detected up to 21 different alleles in 105 individuals. Calculating inbreeding coefficients (F) from the microsatellite allele frequencies gave F ranged between -0.188 to 0.203 with seven positive and two negative values and an average of 0.107 indicating a tolerable degree of population stucture/inbreeding within the proposd founder population.
Sequence diversity
To analyze the Kiyosu population in greater detail, eight mating pairs and their first-generation progeny (i.e. 8 trios) were selected. The genomes of these individuals were profiled using high-throughput short read sequencing, with ~9X mean depth of coverage per line and avriants were identified compared to the Southern Japanese reference HdrR sequence. Approximately 23% of variants detected were monoallelic across Kiyosu, distinguishing this population from the reference HdrR strain. We scored and phased the remaining 77% of variants, giving 4,797,962 SNPs segregating within the Kiyosu population. This polymorphism set provides the opportunity for a far more powerful test of population structure than the microsatellite approach above. Clustering the sample genotype differences by distance metric we found that the parental genotypes were largely equidistant from each other in the tree without visible structure. As expected, the wild individuals clustered closest to each other near the Southern reference strain HdrR collected in the same region of Japan confirming the results from directed genotyping.
LD

LD Decay of Linkage Disequilibrium (LD ) for a sample of Medaka SNPs.
Linkage disequilibrium (LD) is the deviation from independent segregation of alleles between loci, and is dependent on recombination rate and population history. The extent of LD is an important parameter for mapping genotype-phenotype associations within a population. We estimated the LD for the medaka Kiyosu population from the 16 parental sample genotypes expressed as r2, which expresses the key relationship in association mapping. r2 was calculated for all SNP pairs in 1 Mb windows overlapping by 500 kb across Medaka chromosome 1, and plotted against distance for a sample of 21976 pairs. The main plot shows a blow up of distances between 0 and 25Kb, with data for pairs of SNPs up to 1 Mb apart shown in the inset region. Median r2 between pairs of SNPs drops with increasing distance to a minimum around 12.5 kb. In human r2 stabilizes at ~37 kb, ~3 times larger. For our medaka data, ~85% of SNP pairs with r2 > 0.8 mapped to the same gene, with ~37% mapping to the same exon. Thus fine mapping in this Medaka panel should be possible to the resolution of single genes and frequently even to a single exon. We also estimated haplotype block size across 15 of the medaka chromosomes using the method of Gabriel et al. (2002). The mean haplotype block size was 712 bp (median 259 bp) with a maximum of ~78 kb. Example haplotype blocks are available.
Phenotypic Variation
An important feature of a population panel is that it is expected to exhibit appreciable phenotypic variation across individual lines. To characterize one set of phenotypes we took advantage of the Southern inbred lines already in existence since these are likely to be representative of lines generated from the Kiyosu population, allowing us to easily characterize broad sense heritability of phenotypes. We used light microscope-based imaging combined with an automatic annotation algorithm to derive a number of morphometric features across four Southern (HdrR, Icab, HNCMH2, HO5), two Northern strains (Kaga, HNI) and the Kiyosu population (Figure 6A and Supplementary Figures 2 and 3) from both lateral and dorsal viewpoints. The morphometric analysis of the six inbred lines shows reassuringly that the body length measurement was almost perfectly correlated between the two viewpoints. As expected in a fish the majority of the variance between individuals correlates with body length, and so we normalized the remaining morphometric phenotypes by body length. Of the 7 phenotypes analyzed, 4 show greater than 30% broad sense heritability i.e. the differences between strains explained 30% or more of the variance (Figure 6B). An example is shown in Figure 6C where a fish from the HdrR strain has substantially smaller eyes relative to body length than the Icab strain (compare box plot in Figure 6D). The broad sense heritability estimate is similar to analogous morphometric measurements between inbred mouse strains, suggesting that medaka populations have similar levels of phenotypic variation as observed between laboratory mouse strains. Many other established phenotypes that differ between medaka strains have been observed during the prior century of research on this fish (Hyodo-Taguchi 1990; Ishikawa et al. 1999; Kimura et al. 2007) and further work will be needed to examine the suitability of this population for each phenotype. Given the high diversity of alleles in the Southern population we can expect many phenotypes to have least some genetic variance in this population.