 |
PDBsum entry 1hbv

|
|
|
|
|
|

|

|

|

|
|

|
|

|
|

|
|

|
|
|
 |

|

|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Hydrolase (acid protease)
|
PDB id
|
|
|
|
1hbv
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

|

|

|
|
|
|
|
|
|
|
 |
Contents |
 |
|
|
|
|
|
|
|
|
|
|
|
|
* Residue conservation analysis
|
|
|
|
|
|
|
|
|
|
|
Enzyme class 1:
|
|
E.C.2.7.7.-
- ?????
|
|
|
|
|
|
|
Enzyme class 2:
|
|
E.C.2.7.7.49
- RNA-directed Dna polymerase.
|
|
|
|
|
|
|
Reaction:
|
|
DNA(n) + a 2'-deoxyribonucleoside 5'-triphosphate = DNA(n+1) + diphosphate
|
|
|
|
|
|
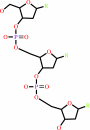 DNA(n)
DNA(n)
|
+
|
2'-deoxyribonucleoside 5'-triphosphate
|
=
|
DNA(n+1)
|
+
|
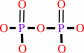 diphosphate
diphosphate
|
|
|
|
|
|
|
|
|
|
Enzyme class 3:
|
|
E.C.2.7.7.7
- DNA-directed Dna polymerase.
|
|
|
|
|
|
|
Reaction:
|
|
DNA(n) + a 2'-deoxyribonucleoside 5'-triphosphate = DNA(n+1) + diphosphate
|
|
|
|
|
|
DNA(n)
|
+
|
2'-deoxyribonucleoside 5'-triphosphate
|
=
|
DNA(n+1)
|
+
|
diphosphate
|
|
|
|
|
|
|
|
|
|
Enzyme class 4:
|
|
E.C.3.1.-.-
|
|
|
|
|
|
|
Enzyme class 5:
|
|
E.C.3.1.13.2
- exoribonuclease H.
|
|
|
|
|
|
|
Reaction:
|
|
Exonucleolytic cleavage to 5'-phosphomonoester oligonucleotides in both 5'- to 3'- and 3'- to 5'-directions.
|
|
|
|
|
|
Enzyme class 6:
|
|
E.C.3.1.26.13
- retroviral ribonuclease H.
|
|
|
|
|
|
|
Enzyme class 7:
|
|
E.C.3.4.23.16
- HIV-1 retropepsin.
|
|
|
|
|
|
|
Reaction:
|
|
Specific for a P1 residue that is hydrophobic, and P1' variable, but often Pro.
|
|
|
|
|
|
|
|
|
Note, where more than one E.C. class is given (as above), each may
correspond to a different protein domain or, in the case of polyprotein
precursors, to a different mature protein.
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
DOI no:
|
J Med Chem
38:3246-3252
(1995)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
A check on rational drug design: crystal structure of a complex of human immunodeficiency virus type 1 protease with a novel gamma-turn mimetic inhibitor.
|
|
S.S.Hoog,
B.Zhao,
E.Winborne,
S.Fisher,
D.W.Green,
R.L.DesJarlais,
K.A.Newlander,
J.F.Callahan,
M.L.Moore,
W.F.Huffman.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
We have previously reported (Newlander et al., J. Med. Chem. 1993, 36,
2321-2331) the design of human immunodeficiency virus type 1 (HIV-1) protease
inhibitors incorporating C7 mimetics that lock three amino acid residues of a
peptide sequence into a gamma-turn. The design of one such compound, SB203238,
was based on X-ray structures of reduced amide aspartyl protease inhibitors. It
incorporates a gamma-turn mimetic in the P2-P1' position, where the carbonyl of
the C7 ring is replaced with an sp3 methylene group yielding a constrained
reduced amide. It shows competitive inhibition with Ki = 430 nM at pH 6.0. The
three-dimensional structure of SB203238 bound to the active site of HIV-1
protease has been determined at 2.3 A resolution by X-ray diffraction and
refined to a crystallographic R-factor (R = sigma magnitude of Fo magnitude of -
magnitude of Fc magnitude of /sigma magnitude of Fo magnitude of, where Fo and
Fc are the observed and calculated structure factor amplitudes, respectively) of
0.177. The inhibitor lies in an extended conformation in the active site;
however, because of the constrained geometry of the C7 ring, it maintains fewer
hydrogen bonds with the protein than in most other HIV-1 protease-inhibitor
complexes. More importantly, the inhibitor binds to the enzyme differently than
predicted in its design, by binding with the P2-P1' alpha-carbon atoms shifted
by approximately one-half a residue toward the N-terminus from their presumed
positions. This study illustrates the importance of structural information in an
approach to rational drug design.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|
Literature references that cite this PDB file's key reference
|
|
|
| |
PubMed id
|
|
Reference
|
|
|
|
|
|
H.B.Thorsteinsdottir,
T.Schwede,
V.Zoete,
and
M.Meuwly
(2006).
How inaccuracies in protein structure models affect estimates of protein-ligand interactions: computational analysis of HIV-I protease inhibitor binding.
|
| |
Proteins,
65,
407-423.
|
|
|
|
|
|
|
M.Cecchini,
P.Kolb,
N.Majeux,
and
A.Caflisch
(2004).
Automated docking of highly flexible ligands by genetic algorithms: a critical assessment.
|
| |
J Comput Chem,
25,
412-422.
|
|
|
|
|
|
|
E.Jenwitheesuk,
and
R.Samudrala
(2003).
Improved prediction of HIV-1 protease-inhibitor binding energies by molecular dynamics simulations.
|
| |
BMC Struct Biol,
3,
2.
|
|
|
|
|
|
|
P.P.Mager
(2001).
The active site of HIV-1 protease.
|
| |
Med Res Rev,
21,
348-353.
|
|
|
|
|
|
|
M.J.Todd,
I.Luque,
A.Velázquez-Campoy,
and
E.Freire
(2000).
Thermodynamic basis of resistance to HIV-1 protease inhibition: calorimetric analysis of the V82F/I84V active site resistant mutant.
|
| |
Biochemistry,
39,
11876-11883.
|
|
|
|
|
|
|
J.D.Tyndall,
and
D.P.Fairlie
(1999).
Conformational homogeneity in molecular recognition by proteolytic enzymes.
|
| |
J Mol Recognit,
12,
363-370.
|
|
|
|
|
|
|
T.J.Stout,
D.Tondi,
M.Rinaldi,
D.Barlocco,
P.Pecorari,
D.V.Santi,
I.D.Kuntz,
R.M.Stroud,
B.K.Shoichet,
and
M.P.Costi
(1999).
Structure-based design of inhibitors specific for bacterial thymidylate synthase.
|
| |
Biochemistry,
38,
1607-1617.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
L.Schaffer,
and
G.M.Verkhivker
(1998).
Predicting structural effects in HIV-1 protease mutant complexes with flexible ligand docking and protein side-chain optimization.
|
| |
Proteins,
33,
295-310.
|
|
|
|
|
|
|
R.B.Rose,
C.S.Craik,
and
R.M.Stroud
(1998).
Domain flexibility in retroviral proteases: structural implications for drug resistant mutations.
|
| |
Biochemistry,
37,
2607-2621.
|
|
|
PDB code:
|
|
|
|
|
|
|
|
T.J.Marrone,
H.Resat,
C.N.Hodge,
C.H.Chang,
and
J.A.McCammon
(1998).
Solvation studies of DMP323 and A76928 bound to HIV protease: analysis of water sites using grand canonical Monte Carlo simulations.
|
| |
Protein Sci,
7,
573-579.
|
|
|
|
|
|
|
J.S.Bardi,
I.Luque,
and
E.Freire
(1997).
Structure-based thermodynamic analysis of HIV-1 protease inhibitors.
|
| |
Biochemistry,
36,
6588-6596.
|
|
|
|
|
|
The most recent references are shown first.
Citation data come partly from CiteXplore and partly
from an automated harvesting procedure. Note that this is likely to be
only a partial list as not all journals are covered by
either method. However, we are continually building up the citation data
so more and more references will be included with time.
Where a reference describes a PDB structure, the PDB
codes are
shown on the right.
|
|


 Links
Links















