 |
PDBsum entry 1a8g

|
|
|
|
|
|

|

|

|

|
|

|
|

|
|

|
|

|
|

|
|
|
 |

|

|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Hydrolase/hydrolase inhibitor
|
PDB id
|
|
|
|
1a8g
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

|

|

|
|
|
|
|
|
|
|
 |
Contents |
 |
|
|
|
|
|
|
|
|
|
|
|
|
* Residue conservation analysis
|
|
|
|
|
|
|
|
|
|
|
Enzyme class 1:
|
|
E.C.2.7.7.-
- ?????
|
|
|
|
|
|
|
Enzyme class 2:
|
|
E.C.2.7.7.49
- RNA-directed Dna polymerase.
|
|
|
|
|
|
|
Reaction:
|
|
DNA(n) + a 2'-deoxyribonucleoside 5'-triphosphate = DNA(n+1) + diphosphate
|
|
|
|
|
|
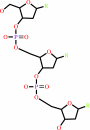 DNA(n)
DNA(n)
|
+
|
2'-deoxyribonucleoside 5'-triphosphate
|
=
|
DNA(n+1)
|
+
|
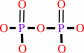 diphosphate
diphosphate
|
|
|
|
|
|
|
|
|
|
Enzyme class 3:
|
|
E.C.2.7.7.7
- DNA-directed Dna polymerase.
|
|
|
|
|
|
|
Reaction:
|
|
DNA(n) + a 2'-deoxyribonucleoside 5'-triphosphate = DNA(n+1) + diphosphate
|
|
|
|
|
|
DNA(n)
|
+
|
2'-deoxyribonucleoside 5'-triphosphate
|
=
|
DNA(n+1)
|
+
|
diphosphate
|
|
|
|
|
|
|
|
|
|
Enzyme class 4:
|
|
E.C.3.1.-.-
|
|
|
|
|
|
|
Enzyme class 5:
|
|
E.C.3.1.13.2
- exoribonuclease H.
|
|
|
|
|
|
|
Reaction:
|
|
Exonucleolytic cleavage to 5'-phosphomonoester oligonucleotides in both 5'- to 3'- and 3'- to 5'-directions.
|
|
|
|
|
|
Enzyme class 6:
|
|
E.C.3.1.26.13
- retroviral ribonuclease H.
|
|
|
|
|
|
|
Enzyme class 7:
|
|
E.C.3.4.23.16
- HIV-1 retropepsin.
|
|
|
|
|
|
|
Reaction:
|
|
Specific for a P1 residue that is hydrophobic, and P1' variable, but often Pro.
|
|
|
|
|
|
|
|
|
Note, where more than one E.C. class is given (as above), each may
correspond to a different protein domain or, in the case of polyprotein
precursors, to a different mature protein.
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
DOI no:
|
J Mol Biol
286:1147-1159
(1999)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
X-ray structure and conformational dynamics of the HIV-1 protease in complex with the inhibitor SDZ283-910: agreement of time-resolved spectroscopy and molecular dynamics simulations.
|
|
S.Ringhofer,
J.Kallen,
R.Dutzler,
A.Billich,
A.J.Visser,
D.Scholz,
O.Steinhauser,
H.Schreiber,
M.Auer,
A.J.Kungl.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
Based on the X-ray structure of the human immunodeficiency virus type-1 (HIV-1)
protease in complex with the statine-derived inhibitor SDZ283-910, a 542 ps
molecular dynamics trajectory was computed. For comparison with the 805 ps
trajectory obtained for the uncomplexed enzyme, the theoretical fluorescence
anisotropy decay of the unliganded protease and the inhibitor complex was
calculated from the trajectories of the Trp6A/Trp6B and Trp42A/Trp42B transition
dipole moments. This enabled us to directly compare the simulated data with the
experimental picosecond time-resolved fluorescence data. Fitting both
experimental and simulated data to the Kohlrausch-Williams-Watts (KWW) function
exp(-t/tauk)beta revealed a very good agreement for the uncomplexed protease as
well as for the SDZ283-910 complex. Binding of the inhibitor induced a faster
decay of both the experimental and the computed protease fluorescence anisotropy
decay. By this integrative approach, the atomic detail of inhibitor-induced
changes in the conformational dynamics of the HIV-1 protease was experimentally
verified and will be used for further inhibitor optimisation.
|
|
|
|
|
|
| |
Selected figure(s)
|
|
|
|
| |
|
|

|
|

|
|
Figure 1.
Figure 1. Structure of the HIV-1 protease/SDZ283-910
inhibitor complex, indicating the position of the inhibitor as
well as of the tryptophan residues.
|
|
Figure 2.
Figure 2. Structure of SDZ283-910. Also included are the
building blocks used for generating the MD input.
|
|
|
|
|
|
| |
The above figures are
reprinted
by permission from Elsevier:
J Mol Biol
(1999,
286,
1147-1159)
copyright 1999.
|
|
| |
Figures were
selected
by an automated process.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|
Literature references that cite this PDB file's key reference
|
 Google scholar
Google scholar
|
|
|
| |
PubMed id
|
|
Reference
|
|
|
|
|
|
T.Lu,
Y.Chen,
and
X.Y.Li
(2010).
An insight into the opening path to semi-open conformation of HIV-1 protease by molecular dynamics simulation.
|
| |
AIDS,
24,
1121-1125.
|
|
|
|
|
|
|
S.Cotesta,
and
M.Stahl
(2006).
The environment of amide groups in protein-ligand complexes: H-bonds and beyond.
|
| |
J Mol Model,
12,
436-444.
|
|
|
|
|
|
|
G.F.Schröder,
U.Alexiev,
and
H.Grubmüller
(2005).
Simulation of fluorescence anisotropy experiments: probing protein dynamics.
|
| |
Biophys J,
89,
3757-3770.
|
|
|
|
|
|
|
V.Helms
(2002).
Electronic excitations of biomolecules studied by quantum chemistry.
|
| |
Curr Opin Struct Biol,
12,
169-175.
|
|
|
|
|
|
|
C.Laboulais,
E.Deprez,
H.Leh,
J.F.Mouscadet,
J.C.Brochon,
and
M.Le Bret
(2001).
HIV-1 integrase catalytic core: molecular dynamics and simulated fluorescence decays.
|
| |
Biophys J,
81,
473-489.
|
|
|
|
|
|
|
P.Furet,
P.Imbach,
P.Fürst,
M.Lang,
M.Noorani,
J.Zimmermann,
and
C.García-Echeverria
(2001).
Modeling of the binding mode of a non-covalent inhibitor of the 20S proteasome. Application to structure-based analogue design.
|
| |
Bioorg Med Chem Lett,
11,
1321-1324.
|
|
|
|
|
|
|
B.Ullrich,
M.Laberge,
F.Tölgyesi,
Z.Szeltner,
L.Polgár,
and
J.Fidy
(2000).
Trp42 rotamers report reduced flexibility when the inhibitor acetyl-pepstatin is bound to HIV-1 protease.
|
| |
Protein Sci,
9,
2232-2245.
|
|
|
|
|
|
The most recent references are shown first.
Citation data come partly from CiteXplore and partly
from an automated harvesting procedure. Note that this is likely to be
only a partial list as not all journals are covered by
either method. However, we are continually building up the citation data
so more and more references will be included with time.
|
|


 Links
Links
















