 |
PDBsum entry 2nxm

|
|
|
|
|
|

|

|

|

|
|

|
|

|
|

|
|

|
|

|
|
|
 |

|

|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Hydrolase/hydrolase substrate
|
PDB id
|
|
|
|
2nxm
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

|

|

|
|
|
|
|
|
|
|
 |
Contents |
 |
|
|
|
|
|
|
|
|
|
|
|
|
* Residue conservation analysis
|
|
|
|
|
|
|
|
|
|
|
Enzyme class 1:
|
|
E.C.2.7.7.-
- ?????
|
|
|
|
|
|
|
Enzyme class 2:
|
|
E.C.2.7.7.49
- RNA-directed Dna polymerase.
|
|
|
|
|
|
|
Reaction:
|
|
DNA(n) + a 2'-deoxyribonucleoside 5'-triphosphate = DNA(n+1) + diphosphate
|
|
|
|
|
|
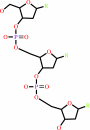 DNA(n)
DNA(n)
|
+
|
2'-deoxyribonucleoside 5'-triphosphate
|
=
|
DNA(n+1)
|
+
|
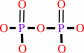 diphosphate
diphosphate
|
|
|
|
|
|
|
|
|
|
Enzyme class 3:
|
|
E.C.2.7.7.7
- DNA-directed Dna polymerase.
|
|
|
|
|
|
|
Reaction:
|
|
DNA(n) + a 2'-deoxyribonucleoside 5'-triphosphate = DNA(n+1) + diphosphate
|
|
|
|
|
|
DNA(n)
|
+
|
2'-deoxyribonucleoside 5'-triphosphate
|
=
|
DNA(n+1)
|
+
|
diphosphate
|
|
|
|
|
|
|
|
|
|
Enzyme class 4:
|
|
E.C.3.1.-.-
|
|
|
|
|
|
|
Enzyme class 5:
|
|
E.C.3.1.13.2
- exoribonuclease H.
|
|
|
|
|
|
|
Reaction:
|
|
Exonucleolytic cleavage to 5'-phosphomonoester oligonucleotides in both 5'- to 3'- and 3'- to 5'-directions.
|
|
|
|
|
|
Enzyme class 6:
|
|
E.C.3.1.26.13
- retroviral ribonuclease H.
|
|
|
|
|
|
|
Enzyme class 7:
|
|
E.C.3.4.23.16
- HIV-1 retropepsin.
|
|
|
|
|
|
|
Reaction:
|
|
Specific for a P1 residue that is hydrophobic, and P1' variable, but often Pro.
|
|
|
|
|
|
|
|
|
Note, where more than one E.C. class is given (as above), each may
correspond to a different protein domain or, in the case of polyprotein
precursors, to a different mature protein.
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
DOI no:
|
Proteins
70:678-694
(2008)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
Computational design and experimental study of tighter binding peptides to an inactivated mutant of HIV-1 protease.
|
|
M.D.Altman,
E.A.Nalivaika,
M.Prabu-Jeyabalan,
C.A.Schiffer,
B.Tidor.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
Drug resistance in HIV-1 protease, a barrier to effective treatment, is
generally caused by mutations in the enzyme that disrupt inhibitor binding but
still allow for substrate processing. Structural studies with mutant, inactive
enzyme, have provided detailed information regarding how the substrates bind to
the protease yet avoid resistance mutations; insights obtained inform the
development of next generation therapeutics. Although structures have been
obtained of complexes between substrate peptide and inactivated (D25N) protease,
thermodynamic studies of peptide binding have been challenging due to low
affinity. Peptides that bind tighter to the inactivated protease than the
natural substrates would be valuable for thermodynamic studies as well as to
explore whether the structural envelope observed for substrate peptides is a
function of weak binding. Here, two computational methods-namely, charge
optimization and protein design-were applied to identify peptide sequences
predicted to have higher binding affinity to the inactivated protease, starting
from an RT-RH derived substrate peptide. Of the candidate designed peptides,
three were tested for binding with isothermal titration calorimetry, with one,
containing a single threonine to valine substitution, measured to have more than
a 10-fold improvement over the tightest binding natural substrate. Crystal
structures were also obtained for the same three designed peptide complexes;
they show good agreement with computational prediction. Thermodynamic studies
show that binding is entropically driven, more so for designed affinity enhanced
variants than for the starting substrate. Structural studies show strong
similarities between natural and tighter-binding designed peptide complexes,
which may have implications in understanding the molecular mechanisms of drug
resistance in HIV-1 protease.
|
|
|
|
|
|
| |
Selected figure(s)
|
|
|
|
| |
|
|

|
|

|
|
Figure 1.
Figure 1. Molecular environments of the Glu-P3 (A) and Thr-P2
(B) peptide residues found to be suboptimal for electrostatic
binding in the RT-RH crystal complex. The position of the P3
glutamate (A, atom colors, center) is pointed away from Arg8B
and makes contact with Phe53A. Calculations suggest an
alternative conformation (yellow) that makes better
electrostatic interactions with Arg8B at the expense of packing.
The wild-type P2 threonine residue (B, center) is situated in a
pocket composed of four hydrophobic residues and makes no polar
interactions. The crystal structure of a designed mutant peptide
with a single threonine-to-valine substitution at the P2
position exhibits a structural rearrangement of the Glu-P3
residue (C, atom colors) as compared to the starting sequence
(A). This rearrangement is well supported by calculation (A, C,
yellow).
|
|
Figure 4.
Figure 4. Superposition of the crystal structures for the RT-RT
peptide (green), Peptide 1 (cyan), Peptide 2 (purple), and
Peptide 3 (yellow) exhibits structural similarity.
|
|
|
|
|
|
| |
The above figures are
reprinted
by permission from John Wiley & Sons, Inc.:
Proteins
(2008,
70,
678-694)
copyright 2008.
|
|
| |
Figures were
selected
by an automated process.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|
Literature references that cite this PDB file's key reference
|
|
|
| |
PubMed id
|
|
Reference
|
|
|
|
|
|
S.Karthik,
and
S.Senapati
(2011).
Dynamic flaps in HIV-1 protease adopt unique ordering at different stages in the catalytic cycle.
|
| |
Proteins,
79,
1830-1840.
|
|
|
|
|
|
|
K.M.Frey,
I.Georgiev,
B.R.Donald,
and
A.C.Anderson
(2010).
Predicting resistance mutations using protein design algorithms.
|
| |
Proc Natl Acad Sci U S A,
107,
13707-13712.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
K.P.Romano,
A.Ali,
W.E.Royer,
and
C.A.Schiffer
(2010).
Drug resistance against HCV NS3/4A inhibitors is defined by the balance of substrate recognition versus inhibitor binding.
|
| |
Proc Natl Acad Sci U S A,
107,
20986-20991.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
R.J.Falconer,
A.Penkova,
I.Jelesarov,
and
B.M.Collins
(2010).
Survey of the year 2008: applications of isothermal titration calorimetry.
|
| |
J Mol Recognit,
23,
395-413.
|
|
|
|
|
|
|
D.J.Mandell,
and
T.Kortemme
(2009).
Computer-aided design of functional protein interactions.
|
| |
Nat Chem Biol,
5,
797-807.
|
|
|
|
|
|
|
S.Chaudhury,
and
J.J.Gray
(2009).
Identification of structural mechanisms of HIV-1 protease specificity using computational peptide docking: implications for drug resistance.
|
| |
Structure,
17,
1636-1648.
|
|
|
|
|
|
The most recent references are shown first.
Citation data come partly from CiteXplore and partly
from an automated harvesting procedure. Note that this is likely to be
only a partial list as not all journals are covered by
either method. However, we are continually building up the citation data
so more and more references will be included with time.
Where a reference describes a PDB structure, the PDB
codes are
shown on the right.
|
|


 Links
Links
















