
|
|
|
|
|
 |
Contents |
 |
|
|
|
|
|
|
|
|
|
|
|


 248 a.a.
248 a.a.
|
|
|
|
|
|
|
|
|
|
|

 271 a.a.
271 a.a.
|
|
|
|
|
|
|
|
|
|
|


 276 a.a.
276 a.a.
|
|
|
|
|
|
|
|
|
|
|

 250 a.a.
250 a.a.
|
|
|
|
|
|
|
|
|
|
|
 255 a.a.
255 a.a.
|
|
|
|
|
|
|
|
|
|
|
 256 a.a.
256 a.a.
|
|
|
|
|
|
|
|
|
* Residue conservation analysis
|
|
|
|
|
|
PDB id:
|
|
|
|
| Name: |
|
Lyase
|
|
|
Title:
|
|
Crystal structure analysis of the 2-enoyl-coa hydratase 2 domain of human peroxisomal multifunctional enzyme type 2
|
|
Structure:
|
|
Peroxisomal multifunctional enzyme type 2. Chain: a, b, c, d, e, f, g, h, i, j, k, l. Fragment: 2-enoyl-coenzyme a hydratase 2 domain. Synonym: mfe-2,d-bifunctional protein, dbp, 17-beta-hydroxysteroid dehydrogenase 4, 17-beta-hsd 4. Engineered: yes. Mutation: yes
|
|
Source:
|
|
Homo sapiens. Human. Organism_taxid: 9606. Gene: hsd17b4, edh17b4. Expressed in: escherichia coli bl21(de3). Expression_system_taxid: 469008.
|
|
Biol. unit:
|
|
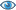 Dimer (from
)
Dimer (from
)
|
|
Resolution:
|
|
|
3.00Å
|
R-factor:
|
0.229
|
R-free:
|
0.265
|
|
|
Authors:
|
|
M.K.Koski,A.M.Haapalainen,J.K.Hiltunen,T.Glumoff
|
Key ref:

|
|
K.M.Koski
et al.
(2005).
Crystal structure of 2-enoyl-CoA hydratase 2 from human peroxisomal multifunctional enzyme type 2.
J Mol Biol,
345,
1157-1169.
PubMed id:
DOI:

|
|
|
Date:
|
|
|
04-Feb-04
|
Release date:
|
15-Feb-05
|
|
|
|
|
|
|
PROCHECK
|

|
|
|
|
|
Headers
|
|
|
|
References
|
|
|
|
|
|

|
|
P51659
(DHB4_HUMAN) -
Peroxisomal multifunctional enzyme type 2 from Homo sapiens
|
|
|
|
Seq:
Struc:
|
|
|
|
736 a.a.
248 a.a.*
|
|

|

|
|
|
|
|
|
|
|
|
|
|
|
|

|
|
P51659
(DHB4_HUMAN) -
Peroxisomal multifunctional enzyme type 2 from Homo sapiens
|
|
|
|
Seq:
Struc:
|
|
|
|
736 a.a.
271 a.a.*
|
|
|

|
|
|
|
|
|
|
|
|
|
|
|
|

|
|
P51659
(DHB4_HUMAN) -
Peroxisomal multifunctional enzyme type 2 from Homo sapiens
|
|
|
|
Seq:
Struc:
|
|
|
|
736 a.a.
276 a.a.*
|
|
|

|
|
|
|
|
|
|
|
|
|
|
|
|

|
|
P51659
(DHB4_HUMAN) -
Peroxisomal multifunctional enzyme type 2 from Homo sapiens
|
|
|
|
Seq:
Struc:
|
|
|
|
736 a.a.
250 a.a.*
|
|
|

|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Enzyme class 2:
|
|
Chains A, B, C, D, E, F, G, H, I, J, K, L:
E.C.1.1.1.n12
- ?????
|
|
|
|
|
|
|
Enzyme class 3:
|
|
Chains A, B, C, D, E, F, G, H, I, J, K, L:
E.C.4.2.1.107
- 3alpha,7alpha,12alpha-trihydroxy-5beta-cholest-24-enoyl-CoA hydratase.
|
|
|
|
|
|
|
Reaction:
|
|
(24R,25R)-3alpha,7alpha,12alpha,24-tetrahydroxy-5beta-cholestan-26-oyl- CoA = (24E)-3alpha,7alpha,12alpha-trihydroxy-5beta-cholest-24-en-26-oyl- CoA + H2O
|
|
|
|
|
|
Enzyme class 4:
|
|
Chains A, B, C, D, E, F, G, H, I, J, K, L:
E.C.4.2.1.119
- enoyl-CoA hydratase 2.
|
|
|
|
|
|
|
Reaction:
|
|
a (3R)-3-hydroxyacyl-CoA = a (2E)-enoyl-CoA + H2O
|
|
|
|
|
|
 (3R)-3-hydroxyacyl-CoA
(3R)-3-hydroxyacyl-CoA
|
=
|
(2E)-enoyl-CoA
|
+
|
H2O
|
|
|
|
|
|
|
|
|
|
|
|
|
Note, where more than one E.C. class is given (as above), each may
correspond to a different protein domain or, in the case of polyprotein
precursors, to a different mature protein.
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
DOI no:
|
J Mol Biol
345:1157-1169
(2005)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
Crystal structure of 2-enoyl-CoA hydratase 2 from human peroxisomal multifunctional enzyme type 2.
|
|
K.M.Koski,
A.M.Haapalainen,
J.K.Hiltunen,
T.Glumoff.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
2-Enoyl-CoA hydratase 2 is the middle part of the mammalian peroxisomal
multifunctional enzyme type 2 (MFE-2), which is known to be important in the
beta-oxidation of very-long-chain and alpha-methyl-branched fatty acids as well
as in the synthesis of bile acids. Here, we present the crystal structure of the
hydratase 2 from the human MFE-2 to 3A resolution. The three-dimensional
structure resembles the recently solved crystal structure of hydratase 2 from
the yeast, Candida tropicalis, MFE-2 having a two-domain subunit structure with
a C-domain complete hot-dog fold housing the active site, and an N-domain
incomplete hot-dog fold housing the cavity for the aliphatic acyl part of the
substrate molecule. The ability of human hydratase 2 to utilize such bulky
compounds which are not physiological substrates for the fungal ortholog, e.g.
CoA esters of C26 fatty acids, pristanic acid and di/trihydroxycholestanoic
acids, is explained by a large hydrophobic cavity formed upon the movements of
the extremely mobile loops I-III in the N-domain. In the unliganded form of
human hydratase 2, however, the loop I blocks the entrance of fatty enoyl-CoAs
with chain-length >C8. Therefore, we expect that upon binding of substrates
bulkier than C8, the loop I gives way, contemporaneously causing a secondary
effect in the CoA-binding pocket and/or active site required for efficient
hydration reaction. This structural feature would explain the inactivity of
human hydratase 2 towards short-chain substrates. The solved structure is also
used as a tool for analyzing the various inactivating mutations, identified
among others in MFE-2-deficient patients. Since hydratase 2 is the last
functional unit of mammalian MFE-2 whose structure has been solved, the
organization of the functional units in the biologically active full-length
enzyme is also discussed.
|
|
|
|
|
|
| |
Selected figure(s)
|
|
|
|
| |
|
|

|
|

|
|
Figure 3.
Figure 3. Ribbon representations of the HsMFE-2(dDhSCP-2LD)
dimer. (a) The upper image shows the four-helix bundle
dimerization motif of hydratase 2 formed by the a-helices a1,
a5, a1' and a5' as well as the two salt bridges formed between
Glu366 and Arg506 (side-chains shown as ball-and-stick
representations). The arrows point to the active sites with
catalytic Asp510 and His515 also shown as magenta. The N and
C-domains are colored as in Figure 2(b). Note that the flexible
loops I-III are fragmented in the lower subunit (subunit J in
the crystal structure of HsMFE-2(dDhSCP-2LD)). The lower image
shows a close-up of the salt bridge Glu366-Arg506. (b) The upper
image shows the dimer after a 90° rotation around the
vertical axis of the upper image of (a). The side-chains of
Asn457 and Tyr347 are shown as ball-and-stick representations,
and the active sites of the human hydratase 2 dimer are
indicated by black arrows. The lower image shows in detail the
interactions of a2 with a1 and with the N-domain b-sheet layer.
The connections shown are described in detail in Discussion.
|
|
Figure 4.
Figure 4. Comparison of the ligand-binding pockets of the
human and C. tropicalis hydratase 2 subunits. (a) Electrostatic
surface potentials of the HsMFE-2(dDhSCP-2LD) ligand-binding
pocket. The positively and negatively charged regions are
colored blue and red, respectively. The residues suggested to
interact with the CoA moiety of the fatty enoyl-CoA substrate
are illustrated. The C-domain overhanging segment is labelled as
the H2-motif. (b) Electrostatic surface potentials of
CtMfe2p(dh[a+b]D) ligand-binding pocket with the bound
(3R)-hydroxydecanoyl-CoA (Protein Data Bank accession code ID
1PN417). The ligand binds to the positively charged CoA-binding
pocket in a bent conformation, where the adenine ring of the
3'-phosphate-ADP moiety points toward the protein, while the
phosphate groups are solvent-exposed. The stronger positive
charge at the surface of the ligand-binding pocket of
CtMfe2p(dh[a+b]D) is created by the side-chains of two lysine
residues (Lys820 and Lys823 of the C-domain overhanging segment)
and an arginine (Arg760 of b-strand b5), which are not found in
HsMFE-2(dDhSCP-2LD). Nevertheless, those residues are not
directly involved in substrate binding. (c) A close-up view of
the CoA-binding pocket after superimposing the apo form of
HsMFE-2(dDhSCP-2LD) (magenta) with the holo form of
CtMfe2p(dh[a+b]D) (light gray). The salt bridge between the
Lys729 of CtMfe2p(dh[a+b]D) and 3'-phosphate of the substrate as
well as the stacking interaction between Arg855 of
CtMfe2p(dh[a+b]D) and adenine ring of the substrate are shown
with black lines. (d) The differences in the region of the
flexible loop I of hydratase 2s from human (green), C.
tropicalis apoenzyme (gray) and C. tropicalis holoenzyme (red)
after superimposition of the three structures. The
(3R)-hydroxydecanoyl-CoA molecule of the C. tropicalis
holoenzyme is also shown. The b-strands b2 and b5 and the
C-domain are only partially shown for clarity. The side-chains
of Met386 and Val404 of HsMFE-2(dDhSCP-2LD) (in pink) as well as
Leu697 of CtMfe2p(dh[a+b]D) (in yellow) are shown. The black
arrow points to the position of the a-methyl group of
branched-chain fatty acids.
|
|
|
|
|
|
| |
The above figures are
reprinted
by permission from Elsevier:
J Mol Biol
(2005,
345,
1157-1169)
copyright 2005.
|
|
| |
Figures were
selected
by an automated process.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|
Literature references that cite this PDB file's key reference
|
|
|
| |
PubMed id
|
|
Reference
|
|
|
|
|
|
J.Jin,
and
U.Hanefeld
(2011).
The selective addition of water to C=C bonds; enzymes are the best chemists.
|
| |
Chem Commun (Camb),
47,
2502-2510.
|
|
|
|
|
|
|
T.J.Haataja,
M.K.Koski,
J.K.Hiltunen,
and
T.Glumoff
(2011).
Peroxisomal multifunctional enzyme type 2 from the fruitfly: dehydrogenase and hydratase act as separate entities, as revealed by structure and kinetics.
|
| |
Biochem J,
435,
771-781.
|
|
|
PDB code:
|
|
|
|
|
|
|
|
G.Möller,
B.Husen,
D.Kowalik,
L.Hirvelä,
D.Plewczynski,
L.Rychlewski,
J.Messinger,
H.Thole,
and
J.Adamski
(2010).
Species used for drug testing reveal different inhibition susceptibility for 17beta-hydroxysteroid dehydrogenase type 1.
|
| |
PLoS One,
5,
e10969.
|
|
|
|
|
|
|
J.M.Crawford,
and
C.A.Townsend
(2010).
New insights into the formation of fungal aromatic polyketides.
|
| |
Nat Rev Microbiol,
8,
879-889.
|
|
|
|
|
|
|
T.Maier,
M.Leibundgut,
D.Boehringer,
and
N.Ban
(2010).
Structure and function of eukaryotic fatty acid synthases.
|
| |
Q Rev Biophys,
43,
373-422.
|
|
|
|
|
|
|
J.M.Crawford,
T.P.Korman,
J.W.Labonte,
A.L.Vagstad,
E.A.Hill,
O.Kamari-Bidkorpeh,
S.C.Tsai,
and
C.A.Townsend
(2009).
Structural basis for biosynthetic programming of fungal aromatic polyketide cyclization.
|
| |
Nature,
461,
1139-1143.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
L.S.Pidugu,
K.Maity,
K.Ramaswamy,
N.Surolia,
and
K.Suguna
(2009).
Analysis of proteins with the 'hot dog' fold: prediction of function and identification of catalytic residues of hypothetical proteins.
|
| |
BMC Struct Biol,
9,
37.
|
|
|
|
|
|
|
S.Jenni,
M.Leibundgut,
D.Boehringer,
C.Frick,
B.Mikolásek,
and
N.Ban
(2007).
Structure of fungal fatty acid synthase and implications for iterative substrate shuttling.
|
| |
Science,
316,
254-261.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
P.Johansson,
A.Castell,
T.A.Jones,
and
K.Bäckbro
(2006).
Structure and function of Rv0130, a conserved hypothetical protein from Mycobacterium tuberculosis.
|
| |
Protein Sci,
15,
2300-2309.
|
|
|
PDB code:
|
|
|
|
|
|
|
|
S.Ferdinandusse,
M.S.Ylianttila,
J.Gloerich,
M.K.Koski,
W.Oostheim,
H.R.Waterham,
J.K.Hiltunen,
R.J.Wanders,
and
T.Glumoff
(2006).
Mutational spectrum of D-bifunctional protein deficiency and structure-based genotype-phenotype analysis.
|
| |
Am J Hum Genet,
78,
112-124.
|
|
|
|
|
|
|
S.Jenni,
M.Leibundgut,
T.Maier,
and
N.Ban
(2006).
Architecture of a fungal fatty acid synthase at 5 A resolution.
|
| |
Science,
311,
1263-1267.
|
|
|
PDB code:
|
|
|
|
|
|
|
|
T.Maier,
S.Jenni,
and
N.Ban
(2006).
Architecture of mammalian fatty acid synthase at 4.5 A resolution.
|
| |
Science,
311,
1258-1262.
|
|
|
PDB code:
|
|
|
|
|
|
|
|
V.Brown,
R.A.Brown,
A.Ozinsky,
J.R.Hesselberth,
and
S.Fields
(2006).
Binding specificity of Toll-like receptor cytoplasmic domains.
|
| |
Eur J Immunol,
36,
742-753.
|
|
|
|
|
|
The most recent references are shown first.
Citation data come partly from CiteXplore and partly
from an automated harvesting procedure. Note that this is likely to be
only a partial list as not all journals are covered by
either method. However, we are continually building up the citation data
so more and more references will be included with time.
Where a reference describes a PDB structure, the PDB
code is
shown on the right.
|

| |


 Links
Links
























