 |
PDBsum entry 1hef

|
|
|
|
|
|

|

|

|

|
|

|
|

|
|

|
|

|
|
|
 |

|

|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Hydrolase/hydrolase inhibitor
|
PDB id
|
|
|
|
1hef
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

|

|

|
|
|
|
|
|
|
|
 |
Contents |
 |
|
|
|
|
|
|
|
|
|
|
|
|
* Residue conservation analysis
|
|
|
|
|
|
|
|
|
|
|
Enzyme class 1:
|
|
E.C.2.7.7.-
- ?????
|
|
|
|
|
|
|
Enzyme class 2:
|
|
E.C.2.7.7.49
- RNA-directed Dna polymerase.
|
|
|
|
|
|
|
Reaction:
|
|
DNA(n) + a 2'-deoxyribonucleoside 5'-triphosphate = DNA(n+1) + diphosphate
|
|
|
|
|
|
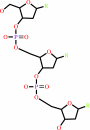 DNA(n)
DNA(n)
|
+
|
2'-deoxyribonucleoside 5'-triphosphate
|
=
|
DNA(n+1)
|
+
|
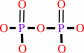 diphosphate
diphosphate
|
|
|
|
|
|
|
|
|
|
Enzyme class 3:
|
|
E.C.2.7.7.7
- DNA-directed Dna polymerase.
|
|
|
|
|
|
|
Reaction:
|
|
DNA(n) + a 2'-deoxyribonucleoside 5'-triphosphate = DNA(n+1) + diphosphate
|
|
|
|
|
|
DNA(n)
|
+
|
2'-deoxyribonucleoside 5'-triphosphate
|
=
|
DNA(n+1)
|
+
|
diphosphate
|
|
|
|
|
|
|
|
|
|
Enzyme class 4:
|
|
E.C.3.1.-.-
|
|
|
|
|
|
|
Enzyme class 5:
|
|
E.C.3.1.13.2
- exoribonuclease H.
|
|
|
|
|
|
|
Reaction:
|
|
Exonucleolytic cleavage to 5'-phosphomonoester oligonucleotides in both 5'- to 3'- and 3'- to 5'-directions.
|
|
|
|
|
|
Enzyme class 6:
|
|
E.C.3.1.26.13
- retroviral ribonuclease H.
|
|
|
|
|
|
|
Enzyme class 7:
|
|
E.C.3.4.23.16
- HIV-1 retropepsin.
|
|
|
|
|
|
|
Reaction:
|
|
Specific for a P1 residue that is hydrophobic, and P1' variable, but often Pro.
|
|
|
|
|
|
|
|
|
Note, where more than one E.C. class is given (as above), each may
correspond to a different protein domain or, in the case of polyprotein
precursors, to a different mature protein.
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
|
J Biol Chem
267:22770-22778
(1992)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
The crystal structures at 2.2-A resolution of hydroxyethylene-based inhibitors bound to human immunodeficiency virus type 1 protease show that the inhibitors are present in two distinct orientations.
|
|
K.H.Murthy,
E.L.Winborne,
M.D.Minnich,
J.S.Culp,
C.Debouck.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
As part of a structure-based drug design program directed against enzyme targets
in the human immunodeficiency virus (HIV), we have determined the
three-dimensional structures of the HIV type 1 protease complexed with two
hydroxyethylene-based inhibitors. The inhibitors (SKF 107457 and SKF 108738) are
hexapeptide substrate analogues with the scissile bond being replaced by a
hydroxyethylene isostere. The structures were determined using x-ray diffraction
data to 2.2 A measured at the Cornell High Energy Synchrotron Source on
hexagonal crystals of each of the complexes. The structures have been
extensively refined using a reciprocal space least-squares method to
conventional crystallographic R factors of 0.186 and 0.159, respectively. The
protein structure differs from that in the unliganded state of the enzyme and is
most similar to that of the structure of the other reported (Jaskolski, M.,
Tomasselli, A. G., Sawyer, T. K., Staples, D. G., Heinrikson, R. L., Schneider,
J., Kent, S. B. H., and Wlodawer, A. (1990) Biochemistry 29, 5889-5907)
hydroxyethylene-based inhibitor complex. Unlike in that structure, however, the
inhibitors are observed, in the present crystal structures, in two equally
abundant orientations that are a consequence of the homodimeric nature of the
enzyme coupled with the asymmetric structures of the inhibitors. Although the
differences between the two inhibitors used in the present study are confined to
the P1' site, the van der Waals interactions made by the inhibitor atoms with
the amino acid residues in the protein differ throughout the structures of the
inhibitors.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|
Literature references that cite this PDB file's key reference
|
|
|
| |
PubMed id
|
|
Reference
|
|
|
|
|
|
D.L.Mobley,
and
K.A.Dill
(2009).
Binding of small-molecule ligands to proteins: "what you see" is not always "what you get".
|
| |
Structure,
17,
489-498.
|
|
|
|
|
|
|
N.Kaushik-Basu,
A.Basu,
and
D.Harris
(2008).
Peptide inhibition of HIV-1: current status and future potential.
|
| |
BioDrugs,
22,
161-175.
|
|
|
|
|
|
|
J.Wang
(2005).
DNA polymerases: Hoogsteen base-pairing in DNA replication?
|
| |
Nature,
437,
E6.
|
|
|
PDB code:
|
|
|
|
|
|
|
|
R.H.Lilien,
B.W.Stevens,
A.C.Anderson,
and
B.R.Donald
(2005).
A novel ensemble-based scoring and search algorithm for protein redesign and its application to modify the substrate specificity of the gramicidin synthetase a phenylalanine adenylation enzyme.
|
| |
J Comput Biol,
12,
740-761.
|
|
|
|
|
|
|
E.Jenwitheesuk,
and
R.Samudrala
(2003).
Improved prediction of HIV-1 protease-inhibitor binding energies by molecular dynamics simulations.
|
| |
BMC Struct Biol,
3,
2.
|
|
|
|
|
|
|
T.Lazaridis,
A.Masunov,
and
F.Gandolfo
(2002).
Contributions to the binding free energy of ligands to avidin and streptavidin.
|
| |
Proteins,
47,
194-208.
|
|
|
|
|
|
|
D.J.Diller,
and
K.M.Merz
(2001).
High throughput docking for library design and library prioritization.
|
| |
Proteins,
43,
113-124.
|
|
|
|
|
|
|
S.Piana,
and
P.Carloni
(2000).
Conformational flexibility of the catalytic Asp dyad in HIV-1 protease: An ab initio study on the free enzyme.
|
| |
Proteins,
39,
26-36.
|
|
|
|
|
|
|
H.M.Krishna Murthy,
K.Judge,
L.DeLucas,
S.Clum,
and
R.Padmanabhan
(1999).
Crystallization, characterization and measurement of MAD data on crystals of dengue virus NS3 serine protease complexed with mung-bean Bowman-Birk inhibitor.
|
| |
Acta Crystallogr D Biol Crystallogr,
55,
1370-1372.
|
|
|
|
|
|
|
R.Brem,
and
K.A.Dill
(1999).
The effect of multiple binding modes on empirical modeling of ligand docking to proteins.
|
| |
Protein Sci,
8,
1134-1143.
|
|
|
|
|
|
|
A.Wlodawer,
and
J.Vondrasek
(1998).
Inhibitors of HIV-1 protease: a major success of structure-assisted drug design.
|
| |
Annu Rev Biophys Biomol Struct,
27,
249-284.
|
|
|
|
|
|
|
R.B.Rose,
C.S.Craik,
and
R.M.Stroud
(1998).
Domain flexibility in retroviral proteases: structural implications for drug resistant mutations.
|
| |
Biochemistry,
37,
2607-2621.
|
|
|
PDB code:
|
|
|
|
|
|
|
|
L.Hong,
A.Treharne,
J.A.Hartsuck,
S.Foundling,
and
J.Tang
(1996).
Crystal structures of complexes of a peptidic inhibitor with wild-type and two mutant HIV-1 proteases.
|
| |
Biochemistry,
35,
10627-10633.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
S.S.Abdel-Meguid
(1993).
Inhibitors of aspartyl proteinases.
|
| |
Med Res Rev,
13,
731-778.
|
|
|
|
|
|
The most recent references are shown first.
Citation data come partly from CiteXplore and partly
from an automated harvesting procedure. Note that this is likely to be
only a partial list as not all journals are covered by
either method. However, we are continually building up the citation data
so more and more references will be included with time.
Where a reference describes a PDB structure, the PDB
code is
shown on the right.
|
|


 Links
Links















