 |
PDBsum entry 2a00

|
|
|
|
|
 |
Contents |
 |
|
|
|
|
|
|
|
|
|
|
* Residue conservation analysis
|
|
|
|
|
|
PDB id:
|
|
|
|
| Name: |
|
Hydrolase
|
|
|
Title:
|
|
The solution structure of the amp-pnp bound nucleotide binding domain of kdpb
|
|
Structure:
|
|
Potassium-transporting atpase b chain. Chain: a. Fragment: kdpbn, nucleotide binding domain of kdpb. Synonym: potassium-transporting p-type atpase, kdpfabc, atp phosphohydrolase [potassium- transporting] b chain, potassium binding and translocating subunit b. Engineered: yes
|
|
Source:
|
|
Escherichia coli. Organism_taxid: 562. Gene: kdpb. Expressed in: escherichia coli bl21(de3). Expression_system_taxid: 469008.
|
|
NMR struc:
|
|
19 models
|
|
|
Authors:
|
|
M.Haupt,M.Bramkamp,M.Coles,K.Altendorf,H.Kessler
|
Key ref:

|
|
M.Haupt
et al.
(2006).
The holo-form of the nucleotide binding domain of the KdpFABC complex from Escherichia coli reveals a new binding mode.
J Biol Chem,
281,
9641-9649.
PubMed id:
DOI:

|
|
|
Date:
|
|
|
15-Jun-05
|
Release date:
|
20-Dec-05
|
|
|
|
|
|
|
PROCHECK
|

|
|
|
|
|
Headers
|
|
|
|
References
|
|
|
|
|
|

|
|
P03960
(KDPB_ECOLI) -
Potassium-transporting ATPase ATP-binding subunit from Escherichia coli (strain K12)
|
|
|
|
Seq:
Struc:
|
|
|
|
682 a.a.
136 a.a.
|
|

|

|
|
|
|
|
|
|
|
|
|
|
|
Key: |
 |
PfamA domain |
|
|
 |
Secondary structure |
|
 |
CATH domain |
|
|
|
|
|
|
|
|
|
|
|
|
|
Enzyme class:
|
|
E.C.7.2.2.6
- P-type K(+) transporter.
|
|
|
|
|
|
|
Reaction:
|
|
K+(out) + ATP + H2O = K+(in) + ADP + phosphate + H+
|
|
|
|
|
|
K(+)(out)
|
+
|
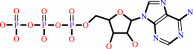 ATP
ATP
|
+
|
H2O
|
=
|
K(+)(in)
Bound ligand (Het Group name = )
matches with 81.25% similarity
|
+
|
 ADP
ADP
|
+
|
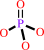 phosphate
phosphate
|
+
|
H(+)
|
|
|
|
|
|
|
|
|
|
Cofactor:
|
|
Mg(2+)
|
|
|
|
|
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
DOI no:
|
J Biol Chem
281:9641-9649
(2006)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
The holo-form of the nucleotide binding domain of the KdpFABC complex from Escherichia coli reveals a new binding mode.
|
|
M.Haupt,
M.Bramkamp,
M.Heller,
M.Coles,
G.Deckers-Hebestreit,
B.Herkenhoff-Hesselmann,
K.Altendorf,
H.Kessler.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
P-type ATPases are ubiquitously abundant enzymes involved in active transport of
charged residues across biological membranes. The KdpB subunit of the
prokaryotic Kdp-ATPase (KdpFABC complex) shares characteristic regions of
homology with class II-IV P-type ATPases and has been shown previously to be
misgrouped as a class IA P-type ATPase. Here, we present the NMR structure of
the AMP-PNP-bound nucleotide binding domain KdpBN of the Escherichia coli
Kdp-ATPase at high resolution. The aromatic moiety of the nucleotide is clipped
into the binding pocket by Phe(377) and Lys(395) via a pi-pi stacking and a
cation-pi interaction, respectively. Charged residues at the outer rim of the
binding pocket (Arg(317), Arg(382), Asp(399), and Glu(348)) stabilize and direct
the triphosphate group via electrostatic attraction and repulsion toward the
phosphorylation domain. The nucleotide binding mode was corroborated by the
replacement of critical residues. The conservative mutation F377Y produced a
high residual nucleotide binding capacity, whereas replacement by alanine
resulted in low nucleotide binding capacities and a considerable loss of ATPase
activity. Similarly, mutation K395A resulted in loss of ATPase activity and
nucleotide binding affinity, even though the protein was properly folded. We
present a schematic model of the nucleotide binding mode that allows for both
high selectivity and a low nucleotide binding constant, necessary for the fast
and effective turnover rate realized in the reaction cycle of the Kdp-ATPase.
|
|
|
|
|
|
| |
Selected figure(s)
|
|
|
|
| |
|
|

|
|

|
|
Figure 7.
FIGURE 7. Zoom into the nucleotide binding pocket. The
stereo view of the binding pocket backs the critical role of
Phe^377 and Lys^395 for nucleotide binding, depicted are all
residues that are within a sphere of 10 Å around the
nucleotide.
|
|
Figure 8.
FIGURE 8. Clip to fit. The schematic picture of the
nucleotide binding pocket illustrates the simple and
energy-saving nucleotide binding mode, enabling the rapid
nucleotide exchange necessary for a functional reaction cycle.
Highlighted are the residues Phe^377, Lys^395, Arg^317, and
Arg^382. Asp^344 is represented as a negative charge at the
bottom of the binding pocket by a red circle.
|
|
|
|
|
|
| |
The above figures are
reprinted
by permission from the ASBMB:
J Biol Chem
(2006,
281,
9641-9649)
copyright 2006.
|
|
| |
Figures were
selected
by the author.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|
Literature references that cite this PDB file's key reference
|
|
|
| |
PubMed id
|
|
Reference
|
|
|
|
|
|
M.G.Palmgren,
and
P.Nissen
(2011).
P-type ATPases.
|
| |
Annu Rev Biophys,
40,
243-266.
|
|
|
|
|
|
|
L.Banci,
I.Bertini,
F.Cantini,
and
S.Ciofi-Baffoni
(2010).
Cellular copper distribution: a mechanistic systems biology approach.
|
| |
Cell Mol Life Sci,
67,
2563-2589.
|
|
|
|
|
|
|
L.Banci,
I.Bertini,
F.Cantini,
S.Inagaki,
M.Migliardi,
and
A.Rosato
(2010).
The binding mode of ATP revealed by the solution structure of the N-domain of human ATP7A.
|
| |
J Biol Chem,
285,
2537-2544.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
T.Tsuda,
and
C.Toyoshima
(2009).
Nucleotide recognition by CopA, a Cu+-transporting P-type ATPase.
|
| |
EMBO J,
28,
1782-1791.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
J.C.Greie,
and
K.Altendorf
(2007).
The K+-translocating KdpFABC complex from Escherichia coli: a P-type ATPase with unique features.
|
| |
J Bioenerg Biomembr,
39,
397-402.
|
|
|
|
|
|
|
S.W.Ginzinger,
F.Gerick,
M.Coles,
and
V.Heun
(2007).
CheckShift: automatic correction of inconsistent chemical shift referencing.
|
| |
J Biomol NMR,
39,
223-227.
|
|
|
|
|
|
The most recent references are shown first.
Citation data come partly from CiteXplore and partly
from an automated harvesting procedure. Note that this is likely to be
only a partial list as not all journals are covered by
either method. However, we are continually building up the citation data
so more and more references will be included with time.
Where a reference describes a PDB structure, the PDB
codes are
shown on the right.
|
|


 Links
Links






