 |
PDBsum entry 2kmx

|
|
|
|
|
 |
Contents |
 |
|
|
|
|
|
|
|
|
|
|
* Residue conservation analysis
|
|
|
|
|
|
|
|
|
|
|
Enzyme class:
|
|
E.C.7.2.2.8
- P-type Cu(+) transporter.
|
|
|
|
|
|
|
Reaction:
|
|
Cu+(in) + ATP + H2O = Cu+(out) + ADP + phosphate + H+
|
|
|
|
|
|
Cu(+)(in)
Bound ligand (Het Group name = )
corresponds exactly
|
+
|
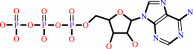 ATP
ATP
|
+
|
H2O
|
=
|
Cu(+)(out)
|
+
|
 ADP
ADP
|
+
|
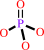 phosphate
phosphate
|
+
|
H(+)
|
|
|
|
|
|
|
|
|
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
DOI no:
|
J Biol Chem
285:2537-2544
(2010)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
The binding mode of ATP revealed by the solution structure of the N-domain of human ATP7A.
|
|
L.Banci,
I.Bertini,
F.Cantini,
S.Inagaki,
M.Migliardi,
A.Rosato.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
We report the solution NMR structures of the N-domain of the Menkes protein
(ATP7A) in the ATP-free and ATP-bound forms. The structures consist of a twisted
antiparallel six-stranded beta-sheet flanked by two pairs of alpha-helices. A
protein loop of 50 amino acids located between beta 3 and beta 4 is disordered
and mobile on the subnanosecond time scale. ATP binds with an affinity constant
of (1.2 +/- 0.1) x 10(4) m(-1) and exchanges with a rate of the order of 1 x
10(3) s(-1). The ATP-binding cavity is considerably affected by the presence of
the ligand, resulting in a more compact conformation in the ATP-bound than in
the ATP-free form. This structural variation is due to the movement of the
alpha1-alpha2 and beta2-beta 3 loops, both of which are highly conserved in
copper(I)-transporting P(IB)-type ATPases. The present structure reveals a
characteristic binding mode of ATP within the protein scaffold of the
copper(I)-transporting P(IB)-type ATPases with respect to the other P-type
ATPases. In particular, the binding cavity contains mainly hydrophobic aliphatic
residues, which are involved in van der Waal's interactions with the adenine
ring of ATP, and a Glu side chain, which forms a crucial hydrogen bond to the
amino group of ATP.
|
|
|
|
|
|
| |
Selected figure(s)
|
|
|
|
| |
|
|

|
|

|
|
Figure 1.
Solution structures of ATP-bound N-MNK (A) and ATP-free N-MNK
(B). The average backbone RMSD between ATP-free and ATP-bound
N-MNK domains is of 1.01 Å. The secondary structure
elements comprise residues 1054–1061 (β1), 1070–1081 (α1),
1090–1100 (α2), 1108–1114 (β2), 1118–1124 (β3),
1178–1183 (β4), 1186–1190 (α3), 1197–1209 (α4),
1212–1218 (β5), and 1221–1230 (β6) for ATP-bound N-MNK and
residues 1054–1061 (β1), 1070–1080 (α1), 1087–1100
(α2), 1110–1114 (β2), 1118–1123 (β3), 1178–1183 (β4),
1186–1191 (α3), 1197–1209 (α4), 1212–1218 (β5), and
1221–1230 (β6) for ATP-free N-MNK, respectively. Top panels,
the radius of the tubes is proportional to the backbone RMSD of
each residue. The unstructured loop was omitted for simplicity.
Bottom panel, the backbone traces for the twenty lowest energy
conformers are superimposed. The unstructured loop is shown in
blue.
|
|
Figure 5.
ATP-binding mode of N-MNK. A, the structure of ATP-bound
N-MNK showing the side chains of hydrophobic amino acids in blue
and of Glu^1081 in yellow, which are in contact with the ATP
molecule. The unstructured loop was omitted for simplicity. In
the zoom on the ATP-binding cavity, the amino acids are labeled.
B, overlay of the structures of ATP-free (pink) and ATP-bound
(red) N-MNK, highlighting the regions of structural variation.
|
|
|
|
|
|
| |
The above figures are
reprinted
by permission from the ASBMB:
J Biol Chem
(2010,
285,
2537-2544)
copyright 2010.
|
|
| |
Figures were
selected
by the author.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|
Literature references that cite this PDB file's key reference
|
|
|
| |
PubMed id
|
|
Reference
|
|
|
|
|
|
M.Bieri,
A.H.Kwan,
M.Mobli,
G.F.King,
J.P.Mackay,
and
P.R.Gooley
(2011).
Macromolecular NMR spectroscopy for the non-spectroscopist: beyond macromolecular solution structure determination.
|
| |
FEBS J,
278,
704-715.
|
|
|
|
|
|
|
N.A.Veldhuis,
M.J.Kuiper,
R.C.Dobson,
R.B.Pearson,
and
J.Camakaris
(2011).
In silico modeling of the Menkes copper-translocating P-type ATPase 3rd metal binding domain predicts that phosphorylation regulates copper-binding.
|
| |
Biometals,
24,
477-487.
|
|
|
|
|
|
|
O.Y.Dmitriev
(2011).
Mechanism of tumor resistance to cisplatin mediated by the copper transporter ATP7B.
|
| |
Biochem Cell Biol,
89,
138-147.
|
|
|
|
|
|
The most recent references are shown first.
Citation data come partly from CiteXplore and partly
from an automated harvesting procedure. Note that this is likely to be
only a partial list as not all journals are covered by
either method. However, we are continually building up the citation data
so more and more references will be included with time.
|
|


 Links
Links
















