DECIPHER v11.40 released
DECIPHER v11.40 is now available, featuring Regional Nonsense Constraint data, EpiSign™ episignatures, and updated NMD predictions.

What’s new in DECIPHER v11.40?
This latest version introduces key improvements including the display of Regional Nonsense Constraint data, EpiSign™ episignatures, updated NMD predictions, and ClinVar aggregate classifications:
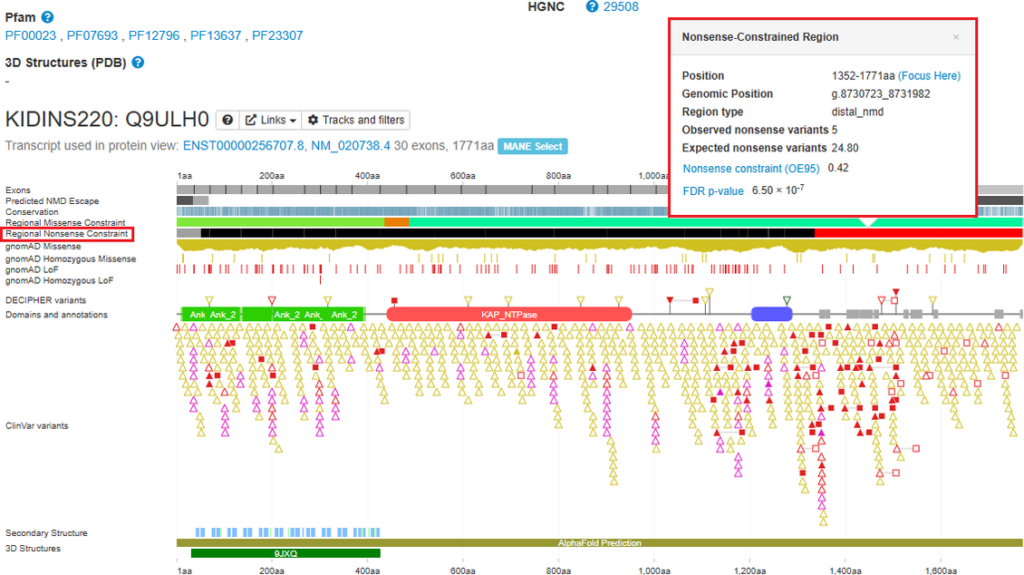
Regional Nonsense Constraint data
Regional Nonsense Constraint data, evidence of intolerance to premature stop codons in the context of nonsense-mediated decay (NMD), can now be viewed on the protein browser. Constraint scores are determined from the number of rare nonsense variants (stop_gained) observed in gnomAD v4.1 exomes, versus the number expected under a null model as described in Blakes et al., 2026. Regions are annotated as constrained, unconstrained or indeterminate (did not meet the criteria for either “constrained” or “unconstrained” annotations). This data assists variant classification by highlighting regions of transcripts that do not tolerant stop_gained variants.
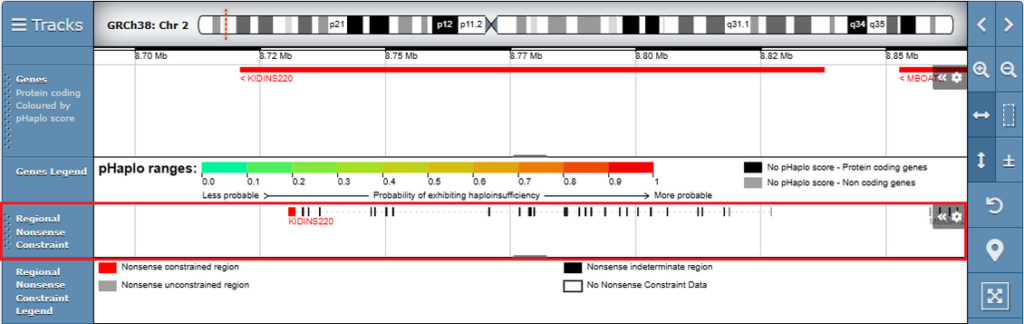
Regional Nonsense Constraint data is also displayed as a track in the genome browser.
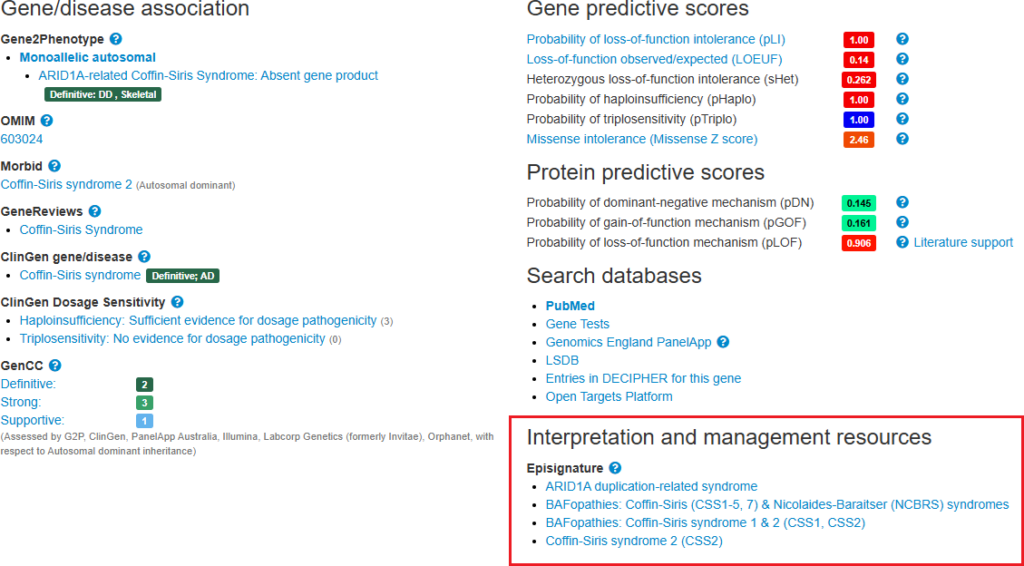
EpiSign™ episignatures
The availability of episignatures by EpiSign™ Inc. are now displayed on gene pages and therapy tabs. Episignatures are reproducible DNA methylation patterns (epigenetic biomarkers) associated with specific genetic disorders or shared biological mechanisms. They provide functional evidence to support variant interpretation and disease classification, and may also assist in the evaluation of patients with suspected rare or neurodevelopmental disorders that remain unresolved following conventional testing approaches. Information regarding clinical laboratories offering EpiSign testing services is available here.
Updated NMD predictions
NMD predictions have been updated on the protein browser. The first 200bp of coding nucleotides are now highlighted as predicted to escape NMD. There is evidence that there is gradual decrease in NMD inhibition between 100-200bp, therefore, the region 100-200bp is shaded in a lighter grey to reflect the decreased NMD inhibition. The PVS1 predictions displayed when classifying a variant using the ACMG/AMP pathogenicity interface, now predicts that if the protein truncating codon falls within the first 200bp of coding nucleotides, the variant is not predicted to undergo NMD.

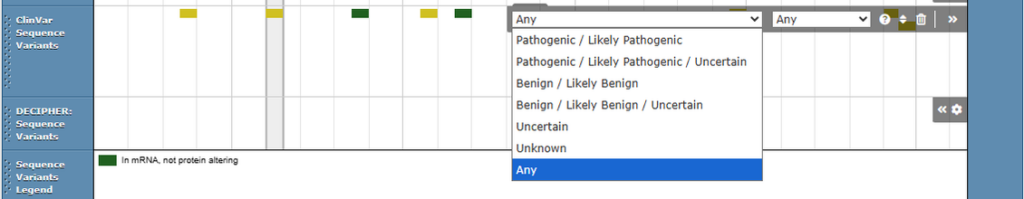
ClinVar aggregate classifications
ClinVar aggregate classifications are now used in the display of ClinVar variants across the site. This includes the colouring of variants (which represents annotated pathogenicity) on the protein browser and filters on the protein and genome browsers.