 |
PDBsum entry 2jqf

|
|
|
|
|
|

|

|

|

|
|

|
|

|
|

|
|
|
 |

|

|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Viral protein
|
PDB id
|
|
|
|
2jqf
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

|

|

|
|
|
|
|
|
|
|
 |
Contents |
 |
|
|
|
|
|
|
|
|
* Residue conservation analysis
|
|
|
|
|
|
|
|
|
|
|
Enzyme class 1:
|
|
E.C.2.7.7.48
- RNA-directed Rna polymerase.
|
|
|
|
|
|
|
Reaction:
|
|
RNA(n) + a ribonucleoside 5'-triphosphate = RNA(n+1) + diphosphate
|
|
|
|
|
|
 RNA(n)
RNA(n)
|
+
|
ribonucleoside 5'-triphosphate
|
=
|
RNA(n+1)
|
+
|
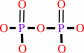 diphosphate
diphosphate
|
|
|
|
|
|
|
|
|
|
Enzyme class 2:
|
|
E.C.3.4.22.28
- picornain 3C.
|
|
|
|
|
|
|
Reaction:
|
|
Selective cleavage of Gln-|-Gly bond in the poliovirus polyprotein. In other picornavirus reactions Glu may be substituted for Gln, and Ser or Thr for Gly.
|
|
|
|
|
|
Enzyme class 3:
|
|
E.C.3.4.22.46
- L-peptidase.
|
|
|
|
|
|
|
Reaction:
|
|
Autocatalytically cleaves itself from the polyprotein of the foot-and-mouth disease virus by hydrolysis of a Lys-|-Gly bond, but then cleaves host cell initiation factor eIF-4G at bonds -Gly-|-Arg- and -Lys-|-Arg-.
|
|
|
|
|
|
Enzyme class 4:
|
|
E.C.3.6.1.15
- nucleoside-triphosphate phosphatase.
|
|
|
|
|
|
|
Reaction:
|
|
a ribonucleoside 5'-triphosphate + H2O = a ribonucleoside 5'-diphosphate + phosphate + H+
|
|
|
|
|
|
ribonucleoside 5'-triphosphate
|
+
|
H2O
|
=
|
ribonucleoside 5'-diphosphate
|
+
|
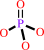 phosphate
phosphate
|
+
|
H(+)
|
|
|
|
|
|
|
|
|
|
|
|
|
Note, where more than one E.C. class is given (as above), each may
correspond to a different protein domain or, in the case of polyprotein
precursors, to a different mature protein.
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
DOI no:
|
J Mol Biol
373:1071-1087
(2007)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
Investigating the substrate specificity and oligomerisation of the leader protease of foot and mouth disease virus using NMR.
|
|
R.Cencic,
C.Mayer,
M.A.Juliano,
L.Juliano,
R.Konrat,
G.Kontaxis,
T.Skern.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
The leader protease (Lbpro) of foot-and-mouth disease virus frees itself during
translation from the viral polyprotein by cleavage between its own C terminus
and the N terminus of the subsequent protein, VP4. Lbpro also specifically
cleaves the host proteins eukaryotic initiation factor (eIF) 4GI and 4GII, thus
disabling host cell protein synthesis. We used NMR to study full-length Lbpro as
well as a shortened species lacking six C-terminal amino acid residues (sLbpro)
to examine the mechanism of self-processing, the quaternary structure and the
substrate specificity. Both Lbpro forms have the same structure in solution as
in the crystal. In the solution structure of sLbpro, the 12 residue C-terminal
extension was flexible and disordered. In contrast, the 18 residue C-terminal
extension of full-length Lbpro was bound by the substrate-binding site of a
neighbouring molecule, resulting in the formation of a stable dimer in solution.
The Lbpro dimer could not be dissociated by increasing the ionic strength or by
dilution. Furthermore, titration with model peptides mimicking the substrates
destabilised the dimer interface without dissociating the dimer. The peptides
were, however, bound by sLbpro in the canonical substrate binding site. Peptide
binding gave rise to chemical shifts of residues around the sLbpro substrate
binding site. Shifts of Asn146 and Glu147 indicated that these residues might
form the enzyme's S1' site and interact with the P1' arginine residue of the
eIF4GI cleavage site. Furthermore, differences in substrate specificity between
sLbpro and Lbpro observed with an in vitro translated protein indicate some
involvement of the C terminus in substrate recognition.
|
|
|
|
|
|
| |
Selected figure(s)
|
|
|
|
| |
|
|

|
|

|
|
Figure 1.
Figure 1. Flexibility of the C-terminal extension (CTE) of
sLb^proC51A. (a) Ensemble of ten sLb^pro NMR structures (shown
in stereo). Regions of regular secondary structure were used for
the coordinate overlay of the final NMR solution structures.
α-Helices are shown in green and β-strands in magenta. The
variability of the structures of the extended CTE region
demonstrates its structural flexibility and presumable lack of
defined structure. (b) ^15N T[2] transverse relaxation times in
ms of sLb^proC51A (blue) and Lb^proC51A (red). Relaxation times
were measured as described in Materials and Methods. The
unstructured CTE is reflected in its T[2] relaxation times being
substantially longer than the globular domain. The roughly
twofold difference in relaxation rates between the two species
implies that Lb^proC51A is a dimer, whereas sLb^proC51A is a
monomer.
|
|
Figure 3.
Figure 3. Solution structure of Lb^pro C51A (a) Comparison of
the dimeric X-ray crystal structure of full-length FMDV Lb^pro
C51A (PDB ID code 1QOL, shown in blue/cyan) with the NMR
solution structure (shown in red/magenta), which is derived from
simple symmetry arguments. Strict enforcement of 2-fold symmetry
between the two halves is required to explain that symmetry of
the measured residual dipolar couplings results in a change of
the relative orientation of the two monomers by about
25–30°. For clarity, the two halves of each monomer are
shown in different colours, and the N and C termini are
labelled. The structures of one half of the dimer (the left
monomers) were overlaid to illustrate the dramatic change of the
position of the other half of the dimer. Residue C133 is shown
as ball-and-stick for orientation. (b) Similar results are
obtained using rigid body simulated annealing refinement with a
limited number of RDCs as orientational restraints and a weak
restraint for the radius of gyration. The bundle of structures
obtained by that procedure is in excellent agreement with the
model derived from simple symmetry reasons shown in (a). The
crystal structure dimer of Lb^pro (PDB ID code 1QOL) is again
shown for reference in blue/cyan. For clarity, the two halves of
each monomer of the structure bundle are shown in different
colours (red/magenta) and the N and C termini are labelled.
Again, the structures of only one half of the dimer (the left
monomers) were overlaid.
|
|
|
|
|
|
| |
The above figures are
reprinted
by permission from Elsevier:
J Mol Biol
(2007,
373,
1071-1087)
copyright 2007.
|
|
| |
Figures were
selected
by an automated process.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|


 Links
Links




















