



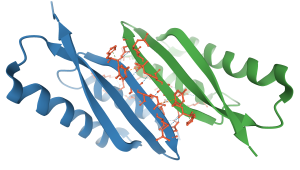

A post-antibiotic era looms

Antimicrobia
PDBe is a founding member of the Worldwide Protein Data Bank (wwPDB) which collects, organises and disseminates data on biological macromolecular structures. wwPDB Partners: RCSB PDB, PDBj, BMRB, EMDB. Read more about PDBe.
| |
New Syntax for PDB DOIs for Entries Released After July 21, 2027
On July 21, 2027, the wwPDB will fully transition to extended 12-character PDB IDs, the PDBx/… |

|
PDB Beta Archive Will Replace Current Archive on July 21, 2027
Starting July 21, 2027: |

|
Updated Validation Reports for Released PDB and EMDB Entries
All validation reports for released PDB and EMDB entries have been updated and are now available… |

|
Poster Prize Awarded at ASBMB
The wwPDB Foundation made an award for an outstanding student presentation at the 2026 meeting… |

