



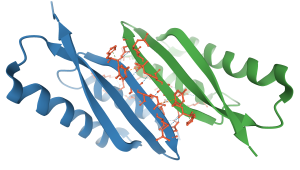

A post-antibiotic era looms

Antimicrobia
PDBe is a founding member of the Worldwide Protein Data Bank (wwPDB) which collects, organises and disseminates data on biological macromolecular structures. wwPDB Partners: RCSB PDB, PDBj, BMRB, EMDB. Read more about PDBe.

|
Milestone: >20,000 Structures Deposited to the PDB in 2025
wwPDB is proud to announce that it surpassed the milestone of 20,000 new structure… |

|
Biocurator Milestone: >10,000 Depositions Processed
Congratulations to biocurator Dr. Deborah Harrus on processing over 10,000 PDB depositions. |

|
Poster Prizes Awarded at The Protein Society
The wwPDB Foundation made awards for outstanding student presentation at the 2025 meeting… |

|
Coming soon: wwPDB Data Processing Distribution Changes
Starting early January 2026, PDBj will biocurate data depositions coming from Oceania in… |

