 |
PDBsum entry 4fbx

|
|
|
|
|
|

|

|

|

|
|

|
|

|
|

|
|

|
|

|
|
|
 |

|

|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Transferase/transferase inhibitor
|
PDB id
|
|
|
|
4fbx
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

|

|

|
|
|
|
|
|
|
PDB id:
|
|
|
|
| Name: |
|
Transferase/transferase inhibitor
|
|
|
Title:
|
|
Complex structure of human protein kinase ck2 catalytic subunit crystallized in the presence of a bisubstrate inhibitor
|
|
Structure:
|
|
Casein kinase ii subunit alpha. Chain: a. Synonym: ck ii alpha. Engineered: yes. Bisubstrate inhibitor. Chain: b. Engineered: yes
|
|
Source:
|
|
Homo sapiens. Human. Organism_taxid: 9606. Gene: csnk2a1, ck2a1. Expressed in: escherichia coli. Expression_system_taxid: 562. Synthetic: yes
|
|
Resolution:
|
|
|
2.33Å
|
R-factor:
|
0.202
|
R-free:
|
0.249
|
|
|
Authors:
|
|
E.Enkvist,K.Viht,N.Bischoff,J.Vahter,S.Saaver,G.Raidaru,O.- G.Issinger,K.Niefind,A.Uri
|
|
Key ref:
|
|
E.Enkvist
et al.
(2012).
A subnanomolar fluorescent probe for protein kinase CK2 interaction studies.
Org Biomol Chem,
10,
8645-8653.
PubMed id:
|
|
|
Date:
|
|
|
23-May-12
|
Release date:
|
17-Oct-12
|
|
|
|
|
|
|
PROCHECK
|

|
|
|
|
|
Headers
|
|
|
|
References
|
|
|
|
|
|

|
|
P68400
(CSK21_HUMAN) -
Casein kinase II subunit alpha from Homo sapiens
|
|
|
|
Seq:
Struc:
|
|
|
|
391 a.a.
333 a.a.
|
|

|

|

|
|
|
|
|
|
|
|
|
|
|
Key: |
 |
PfamA domain |
|
|
 |
Secondary structure |
|
 |
CATH domain |
|
|
|
|
|
|
|
|
|
|
|
|
|
Enzyme class:
|
|
E.C.2.7.11.1
- non-specific serine/threonine protein kinase.
|
|
|
|
|
|
|
Reaction:
|
|
|
1.
|
L-seryl-[protein] + ATP = O-phospho-L-seryl-[protein] + ADP + H+
|
|
2.
|
L-threonyl-[protein] + ATP = O-phospho-L-threonyl-[protein] + ADP + H+
|
|
|
|
|
|
|
L-seryl-[protein]
|
+
|
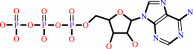 ATP
ATP
|
=
|
O-phospho-L-seryl-[protein]
|
+
|
 ADP
ADP
|
+
|
H(+)
|
|
|
|
|
|
|
L-threonyl-[protein]
|
+
|
ATP
|
=
|
O-phospho-L-threonyl-[protein]
|
+
|
ADP
|
+
|
H(+)
|
|
|
|
|
|
|
|
|
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
|
Org Biomol Chem
10:8645-8653
(2012)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
A subnanomolar fluorescent probe for protein kinase CK2 interaction studies.
|
|
E.Enkvist,
K.Viht,
N.Bischoff,
J.Vahter,
S.Saaver,
G.Raidaru,
O.G.Issinger,
K.Niefind,
A.Uri.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
Up-regulation of an acidophilic protein kinase, CK2, has been established in
several types of cancer. This cognition has made CK2 an important target for
drug development for cancer chemotherapy. The characterization of potential drug
candidates, determination of the structure and clarification of the functions of
CK2 could be facilitated by the application of small-molecule fluorescent probes
that bind to the active site of the enzyme with high affinity and selectivity.
We have used a bisubstrate approach for the development of a highly potent
inhibitor of CK2. 4,5,6,7-Tetrabromo-1H-benzimidazole was conjugated with
peptides containing multiple aspartate residues via different linkers. The
design of the inhibitors was by crystallographic analysis of the complex of an
inhibitor with the catalytic subunit of the enzyme (CK2α). The inhibitory
potency of the synthesized compounds was established in a kinetic assay that
used thin layer chromatography for the measurement of the rate of
phosphorylation of fluorescently labelled peptide 5-TAMRA-RADDSDDDDD. The most
potent inhibitor, ARC-1502 (K(i) = 0.5 nM), revealed high selectivity for CK2α
in a panel of 140 protein kinases. Labelling of ARC-1502 with PromoFluor-647
gave the fluorescent probe ARC-1504 that possessed subnanomolar affinity towards
both CK2α and the holoenzyme. The probe was used in a fluorescence
anisotropy-based binding assay to measure the concentration of CK2α and
characterize non-labelled ligands binding to the active site of CK2α.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|



 Links
Links




