 |
PDBsum entry 1pn0

|
|
|
|
|
|

|

|

|

|
|

|
|

|
|

|
|

|
|

|
|

|
|
|
 |

|

|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Oxidoreductase
|
PDB id
|
|
|
|
1pn0
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

|

|

|
|
|
|
|
|
|
|
 |
Contents |
 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
* Residue conservation analysis
|
|
|
|
|
|
|
|
|
|
|
Enzyme class:
|
|
E.C.1.14.13.7
- phenol 2-monooxygenase (NADPH).
|
|
|
|
|
|
|
Reaction:
|
|
phenol + NADPH + O2 + H+ = catechol + NADP+ + H2O
|
|
|
|
|
|
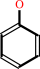 phenol
phenol
|
+
|
 NADPH
NADPH
|
+
|
 O2
O2
|
+
|
H(+)
Bound ligand (Het Group name = )
corresponds exactly
|
=
|
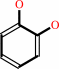 catechol
catechol
|
+
|
 NADP(+)
NADP(+)
|
+
|
H2O
|
|
|
|
|
|
|
|
|
|
Cofactor:
|
|
FAD
|
|
|
|
|
|
FAD
Bound ligand (Het Group name =
FAD)
corresponds exactly
|
|
|
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
DOI no:
|
Acta Crystallogr D Biol Crystallogr
59:1597-1602
(2003)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
High-resolution structure of phenol hydroxylase and correction of sequence errors.
|
|
C.Enroth.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
The crystal structure model of phenol hydroxylase has been corrected for 11
sequence errors and refined against new data to 1.7 A resolution. The higher
resolution data together with careful exploitation of non-crystallographic
symmetry restraints and the use of many small groups for refinement of
anisotropic displacement parameters resulted in a large decrease in the
crystallographic R factor. The final crystallographic free R factor is 18.0%,
which should be compared with the values of 27.8% for the previously published
model (PDB code 1foh). The rebuilding and re-refinement procedure is described.
A comparison with the previously published model was performed and possible
biochemical implications are discussed. No large differences suggesting gross
errors in the earlier model were found. The actual differences between these two
models give an indication of the level of ambiguity and inaccuracy that may be
found in a well refined protein model at 2.4 A resolution.
|
|
|
|
|
|
| |
Selected figure(s)
|
|
|
|
| |
|
|

|
|
Figure 2.
Figure 2 A figure showing the eight structural domains of a
subunit which were treated as independent domains in the TLS
refinement. FAD, which was treated as another separate TLS
domain, is shown in ball-and-stick representation. This figure
was prepared with MOLSCRIPT (Kraulis, 1991[Kraulis, P. J.
(1991). J. Appl. Cryst. 24, 946-950.]) and Raster3D (Merritt &
Murphy, 1994[Merritt, E. A. & Murphy, M. E. P. (1994). Acta
Cryst. D50, 869-873.]).
|
|
|
|
|
|
| |
The above figure is
reprinted
by permission from the IUCr:
Acta Crystallogr D Biol Crystallogr
(2003,
59,
1597-1602)
copyright 2003.
|
|
| |
Figure was
selected
by an automated process.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|
Literature references that cite this PDB file's key reference
|
|
|
| |
PubMed id
|
|
Reference
|
|
|
|
|
|
W.Nishima,
G.Qi,
S.Hayward,
and
A.Kitao
(2009).
DTA: dihedral transition analysis for characterization of the effects of large main-chain dihedral changes in proteins.
|
| |
Bioinformatics,
25,
628-635.
|
|
|
|
|
|
|
K.Ida,
M.Kurabayashi,
M.Suguro,
Y.Hiruma,
T.Hikima,
M.Yamomoto,
and
H.Suzuki
(2008).
Structural basis of proteolytic activation of L-phenylalanine oxidase from Pseudomonas sp. P-501.
|
| |
J Biol Chem,
283,
16584-16590.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
D.P.Ballou
(2007).
Crystallography gets the jump on the enzymologists.
|
| |
Proc Natl Acad Sci U S A,
104,
15587-15588.
|
|
|
|
|
|
|
K.S.Ryan,
A.R.Howard-Jones,
M.J.Hamill,
S.J.Elliott,
C.T.Walsh,
and
C.L.Drennan
(2007).
Crystallographic trapping in the rebeccamycin biosynthetic enzyme RebC.
|
| |
Proc Natl Acad Sci U S A,
104,
15311-15316.
|
|
|
PDB codes:
|
|
|
|
|
|
|
The most recent references are shown first.
Citation data come partly from CiteXplore and partly
from an automated harvesting procedure. Note that this is likely to be
only a partial list as not all journals are covered by
either method. However, we are continually building up the citation data
so more and more references will be included with time.
Where a reference describes a PDB structure, the PDB
codes are
shown on the right.
|
|



 Links
Links














