 |
PDBsum entry 4o8i
|
|

|
|
PDB id:
|
|
|
|
| Name: |
|
Isomerase
|
|
|
Title:
|
|
1.45a resolution structure of peg 400 bound cyclophilin d
|
|
Structure:
|
|
Peptidyl-prolyl cis-trans isomerase f, mitochondrial. Chain: a. Synonym: ppiase f, cyclophilin d, cyp-d, cypd, cyclophilin f, mitochondrial cyclophilin, cyp-m, rotamase f. Engineered: yes. Mutation: yes
|
|
Source:
|
|
Homo sapiens. Human. Organism_taxid: 9606. Gene: ppif, cyp3. Expressed in: escherichia coli. Expression_system_taxid: 469008.
|
|
Resolution:
|
|
|
1.45Å
|
R-factor:
|
0.161
|
R-free:
|
0.190
|
|
|
Authors:
|
|
S.Lovell,K.R.Valasani,K.P.Battaile,C.Wang,S.S.Yan
|
|
Key ref:
|
|
K.R.Valasani
et al.
(2014).
High-resolution crystal structures of two crystal forms of human cyclophilin D in complex with PEG 400 molecules.
Acta Crystallogr F Struct Biol Commun,
70,
717-722.
PubMed id:
DOI:
|
|
|
Date:
|
|
|
27-Dec-13
|
Release date:
|
11-Jun-14
|
|
|
|
|
|
|
PROCHECK
|

|
|
|
|
|
Headers
|
|
|
|
References
|
|
|
|
|
|

|
|
P30405
(PPIF_HUMAN) -
Peptidyl-prolyl cis-trans isomerase F, mitochondrial from Homo sapiens
|
|
|
|
Seq:
Struc:
|
|
|
|
207 a.a.
164 a.a.*
|
|

|

|
|
|
|
|
|
|
|
|
|
|
|
Key: |
 |
PfamA domain |
|
|
 |
Secondary structure |
|
 |
CATH domain |
|
|
*
PDB and UniProt seqs differ
at 1 residue position (black
cross)
|
|
|
|
|
|
|
|
|
|
|
|
|
Enzyme class:
|
|
E.C.5.2.1.8
- peptidylprolyl isomerase.
|
|
|
|
|
|
|
Reaction:
|
|
[protein]-peptidylproline (omega=180) = [protein]-peptidylproline (omega=0)
|
|
|
|
|
|
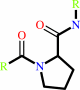 Peptidylproline (omega=180)
Peptidylproline (omega=180)
|
=
|
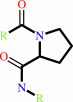 peptidylproline (omega=0)
peptidylproline (omega=0)
|
|
|
|
|
|
|
|
|
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
DOI no:
|
Acta Crystallogr F Struct Biol Commun
70:717-722
(2014)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
High-resolution crystal structures of two crystal forms of human cyclophilin D in complex with PEG 400 molecules.
|
|
K.R.Valasani,
E.A.Carlson,
K.P.Battaile,
A.Bisson,
C.Wang,
S.Lovell,
S.ShiDu Yan.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
Cyclophilin D (CypD) is a key mitochondrial target for amyloid-β-induced
mitochondrial and synaptic dysfunction and is considered a potential drug target
for Alzheimer's disease. The high-resolution crystal structures of primitive
orthorhombic (CypD-o) and primitive tetragonal (CypD-t) forms have been
determined to 1.45 and 0.85 Å resolution, respectively, and are nearly
identical structurally. Although an isomorphous structure of CypD-t has
previously been reported, the structure reported here was determined at atomic
resolution, while CypD-o represents a new crystal form for this protein. In
addition, each crystal form contains a PEG 400 molecule bound to the same region
along with a second PEG 400 site in CypD-t which occupies the cyclosporine A
inhibitor binding site of CypD. Highly precise structural information for CypD
should be extremely useful for discerning the detailed interaction of small
molecules, particularly drugs and/or inhibitors, bound to CypD. The 0.85 Å
resolution structure of CypD-t is the highest to date for any CypD structure.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|


 Links
Links




