 |
The PDBe search database
History
The Macromolecular Structure Group (MSD) is the European project for the collection, management and distribution of data about macromolecular structures. The PDBe has the aim to serve as an alternative complementary and extensible database derived in part from the Protein Data Bank (PDB) and operating under the wwPDB international collaboration.
PDBe and others have recognised long ago the limitations of the PDB flat file format and the need of an extensible framework for macromolecular structure related information.
After taking into account the advances in information management and database technologies over the last decade, PDBe adopted the pragmatic approach of using relational databases in order to support its operations.
The initial step was to develop an internal database that would help with the processing of new PDB entries. This database the "deposition database" is designed following normalisation principles in order to enforce data consistency. After loading, the "consistent" data are exported back to PDB flat files and introduced in the wwPDB repository.
The next step was to use relational database technology in order to offer web services that would allow the external users a toolset for searching and using the PDBe work.
The "deposition database" is now not fit for purpose.
The focus on a "normalised" design has always to come in expense of
simplicity, ease of use and performance.
This is often solved by transforming the main archive database to another "data warehouse" database that will de-normalise, aggregate and simplify it. This is exactly the MSDSD (PDBe search database).
It soon became obvious that this database could also serve users that would like to access it directly - even get a replica copy - to use it an alternative to PDB flat files.
In that way the could use all the available tools and technologies that are available for relational databases and utilise the power and flexibility of relational database technology and SQL.
The MSDSD
Is a rigid relational database which is a
- Simplified,
- Restructured and
- Enriched
reorganisation of the internal PDBe Deposition database.
The Deposition database itself is used to
-
Process new PDB depositions
-
Facilitate the task of data clean up,
The idea behind the PDBe search database is to provide a fast and easy to use public
relational database for
-
Flexible search and data retrieval
-
Cleaned up data
-
For example hundreds of 3 letter codes (like D1P in entries 1g5d, 1g5e)
are useless duplicates of existing ones like ORP and in cases like these
the 3 letter code D1P has become obsolete and replaced by ORP
-
In thousands other cases the atom names in PDB ATOM lines, do not match
with other entries, for the same 3 letter code.
-
Facilitate knowledge discovery
By providing consistently derived information for the complete PDB
like
- Secondary structure
- Quaternary structure of the biological entity
- Active sites
- Classification cross-references
By providing summarised information to improve performance for data
analysis and data mining operations
-
Integrate with external databases and provide
cross-references in a uniform and consistent way.
-
Is always up-to date, synchronised weekly with the repository deposition
database
How to access and use MSDSD
We expect that the majority of the users will use MSDSD indirectly by accessing some of our online search services available from our website. All these services depend and use the production MSDSD database that we maintain and update on a regular (weekly) basis. Most of these services also depend on several other internal optimisation structures and components that are not part of the MSDSD core. For this, we do not always intend to offer them as a package that one would be able to download and run locally.
For more demanding users of the MSDSD database we have several options for using directly relational operations on MSDSD.
The idea is that these users may take advantage of the power and flexibility of database technology in order to utilise the MSDSD in novel ways,
and also built on it or extend it independently.
The choice of which option to use will depend on the needs and resources such as:
-
Available hardware:
-
Maintenance:
-
Programming:
-
Heavy usage:
-
Availability:
-
Extensibility:
-
Up-to-date:
Below there is a summary of the 4 available options we support for using relational operations and SQL
directly and their strong (green), not-so-strong(orange), and weak (red) points.
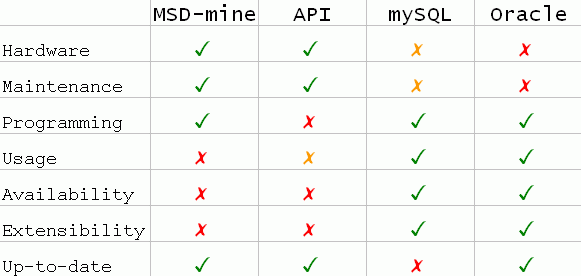
MSD-API and MSD-mine
These are online services that offer direct SQL execution over the web. Both these services will impose limitations
on what an individual user may do and the resources (database CPU time, temporary disk space etc) he may use. This is done in order to avoid over-demanding
requests that would degrade the availability and performance of our local databases.
Users that are not satisfied by what these services offer will have to replicate a local copy of MSDSD using one of the other
options described below.
In brief MSD-mine is a web application for interactive exploration of MSDSD. It allows users to interactively build
arbitrary queries over MSDSD that can then be also used for interactive data-analysis and data mining. Its main aim is to familiarise users
with the MSDSD data.
The MSD-API web service enables developers to query the MSDSD directly from their own application programs in their favourite environment - such as Java, C/C++, Perl using technologies like SOAP and WSDL and is based on Distributed Computing and Grid concepts. The MSD-API offers the full power and flexibility of ad-hoc SQL but needs programming and SQL skills and is available for registered users.
For more information and details follow the corresponding links given above.
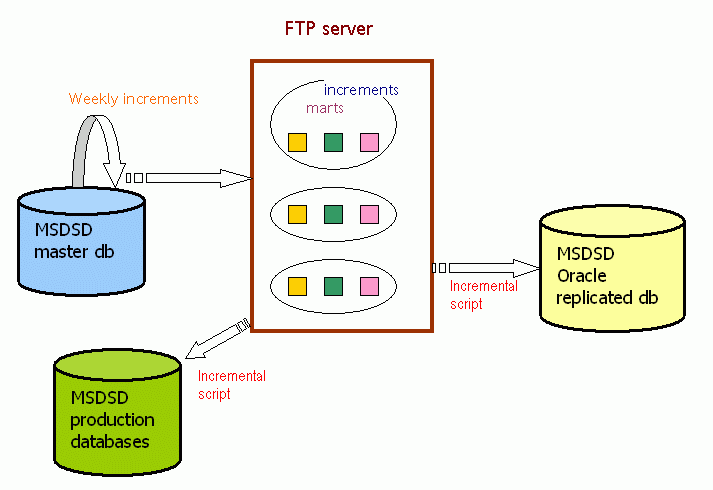
Replication on Oracle
MSDSD is free for academic research and can be downloaded from our ftp site.
To obtain a license, please fill an application form and post three copies to:
Dr Melford John
Database administrator
Macromolecular Database Structure
European Bioinformatics Institute
Welcome Trust Genome Campus
Hinxton, Cambridge, CB10 1SD
United Kingdom
This is the most advanced remote replication option that we offer. It is available for registered users that fill in and post a free of charge
MSDSD license document.
It uses one of the most advanced and powerful commercial relational database servers and is the option that we recommend for the more
serious users of MSDSD and our collaborators. Additionally since we also use it at MSD, we are able to offer more support and advice.
For the Oracle replication option we also offer frequent (weekly) increments for users that wish to follow closely the evolution of our local master MSDSD and of the PDB.
The disadvantages of this option are that users will need to have an oracle server license, some database administration support and adequate hardware infrastructure.
Typically a user of this replication will download and install the latest full release (full transformation) of MSDSD using the full installation instructions. Such full releases take place on a sparse (yearly) basis, and this is the time of MSDSD reconciliation, since all PDB entries are refreshed and creeping inconsistencies are resolved.
In the meantime between releases (full transformations) the user may run the automatic synchronisation script (typically set in a crontab) that will allow the download and inclusion of increments for the new PDB entries that are released every week.
Any corrections in reference data will not propagate back to the affected old entries in order to keep the increments manageable, so the only time that the full set of MSDSD relational constraints is guaranteed, is only immediately after a full release.
The MSDSD and the incremental updates are organised in sections ("marts") so users are free to install and increment, just the marts that they are interested in. There is also the option to specify which tables of a mart a user wishes to have installed, so users may in general replicate just a few individual tables.
For more information you may contact the PDBe group
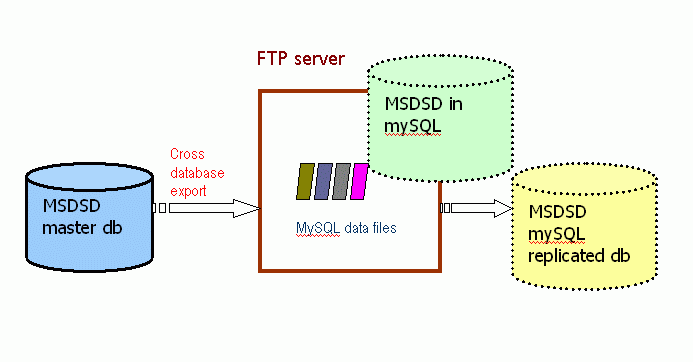
Replication on mySQL
This is the alternative open source database replication that we offer. We have chosen to support mySQL instead of other similar alternatives, because at the time it seems to be the easier to install and start with. It also has the fewer platform dependencies and requires almost no system administrator involvement in order to set-up. The idea is to offer something that will require the minimum effort to install and give it a try for a researcher who is not an expert in the IT area and has no dedicated resources and support.
It should be easy to replicate even on a normal desktop workstation with a fair amount of disk space.
It also does not bind the user community to a commercial software database vendor.
The disadvantages of this options are that mySQL may not always have the sophistication and speed of a commercial database (for complex queries), we do not offer frequent incrementals and that we do not use it much, so we will not be able to offer as much support and advice.
It is also available for registered users that fill in and post a free of charge MSDSD mySQL license document (in 3 copies).
Typically a user of this replication will download and install directly the mySQL data-files of the tables he is interested in from our FTP server following the mySQL installation instructions. The tables are available in compressed myIsam format without any pre-built indexes.
For more information you may contact the PDBe group
MSDSD and flat files (PDB, mmCif, XML)
A frequently asked question about MSDSD is why the database is not available in XML and other
flat file formats (XML,mmCif,clean-up PDB).
The reason is that we feel that XML and other flat-files have to be based on a
rigid and systematic standardisation in order to be useful.
This work is done as part of the wwPDB collaboration and we would advice users to
refer to the wwPDB
-
MSDSD is based on relational database technology and it is available
on relational database technology. It is not a collection of flat files.
-
It is not another data store for the PDB and does not intend
to replace the PDB
-
While it is partially based on mmCif terminology and organisation,
it is not bound to use it when this is unsuitable
-
It includes complete and consistent sections of automatically or semi-automatically
derived information that are not part of the PDB
-
It includes complete and consistent cross-references as well as
reference information from external databases. All these are not part of
the PDB
-
It focuses mainly on "Assemblies", the quaternary structure that
corresponds to the actual biological entity, as the starting point for
determining the actual structural characteristics of proteins
MSDSD conventions
MSDSD (with some exceptions) is following a standard set of conventions in its design and architecture.
Some understanding of these conventions will help anyone interested in learning the
MSDSD schema, regardless of the method he chooses to use in order to use it (replication, API).
For a more systematic study you will have also to consult the MSDSD reference documentation.
- We use user friendly and meaningful names that are familiar to the end users instead of precise names. We will use the names CHAIN and RESIDUE instead of ENTITY_INSTANCE or COMPONENT even though that the entities that they model are not always very strictly speaking chains or residues (i.e. water groups and waters or bound molecules). This means diversion in some cases from the strict mmCif terms.
- We will follow de facto standards wherever there is no necessity to oppose them. For example we will use the PDB nomenclature and ordering for atoms and PDB style names for chains.
- Table names in sections of the model may be marked up with a common prefix (i.e. "CHEM" like in CHEM_COMP,CHEM_ATOM)
- Tables will have an abstract identifier attribute that will be used in order to implement foreign keys. Only these attributes should end with the "ID" suffix.
- The abstract identifiers are not guaranteed to remain constant in different MSDSD releases. There is always another set of naming identifiers (like ACCESSION_CODE,ASSEMBLY_SERIAL etc) for every table and these should be used by external users in order to refer to MSDSD records.
- The abstract identifier attributes of entities should have the same name as the entity and the "ID" suffix i.e. CHEM_ATOM_ID for CHEM_ATOM.
- Attributes that are external identifiers will not have the ID suffix and will be named to the CODE suffix (i.e. CIF_CODE) if they are alphanumeric, to the NO suffix (i.e. EBI_NO) when they are numeric, or the the SERIAL suffix when they are serial numbers (i.e. ASSEMBLY_SERIAL)
- Attributes that are "propagated" (denormalised) like foreign keys or foreign names should keep the same names as in the parent tables (i.e. CHEM_COMP_ID and CHEM_COMP_CODE on CHEM_ATOM) or use a consistent role name (i.e. SUPERCEDED_BY_COMP_ID - SUPERCEDED_BY_COMP_CODE).
- Usage of database or other language keywords is avoided (i.e. CLASS,TYPE,GROUP,CODE,TEXT). Table and column names should include alphanumeric characters only and the underscore (not #,$ etc)
- Names though are kept as short as possible
- Legacy names and identifiers are available where appropriate and have corresponding prefixes (i.e. PDB_CODE). Their uniqueness is not guaranteed and in most cases cannot be used as keys for reference
- Cryptic names and abbreviations are avoided i.e. NUM_RESIDUES instead of N_RESID and RELEASE_STATUS instead of REL_STATUS
- If this is not possible or in cases where attribute names are too vague: ID, CLASS, TYPE, CODE, ORDER they are prefixed with the name of the table (i.e. CHAIN_CODE, CHEM_BOND_ORDER). In cases that these attributes are propagated, the prefix is not duplicated (i.e. not CHAIN_CHAIN_CODE). The entity name should not be used as a prefix in any other case. Especially the fact that a column will be propagated should not affect the decision to prefix it with the table name.
- Boolean attributes get values 'Y'/'N' and have "FLAG" as suffix
- Counts start with the NUM prefix i.e. NUM_RESIDUES
- Extending the model with new attributes or entities is not a problem and has to be expected. Modification or removal of attributes or entities though, is strongly avoided. Inclusion of unimplemented "empty" attributes for future use is carefully considered.
- There may be a set of internal cryptic database identifier attributes usually with the INT prefix (i.e. INT_DEIC_ID) and these should not be used by external users
Sections of the PDBe search database
The PDBe search database is organised in
interrelated sections. Some of these sections are in the centre
of the database, while others may be decoupled and ignored for those
that are not interested in them.
-
Ligands
This is a consistent and enriched library of ligands, small molecules
and monomers that are referred by each residue and atom. There is complete
and consistent reference information for any small molecule and aminoacid
like for example CPM
 that includes detailed
information about its atoms and bonds, their standard nomenclature and
ordering, as well as their important characteristics like aromaticity and
stereochemistry. Any atom or residue in any actual structure, that does
not include and follow a reference in an atom or ligand of this dictionary,
is simply unidentified and requires cleanup. that includes detailed
information about its atoms and bonds, their standard nomenclature and
ordering, as well as their important characteristics like aromaticity and
stereochemistry. Any atom or residue in any actual structure, that does
not include and follow a reference in an atom or ligand of this dictionary,
is simply unidentified and requires cleanup.
-
Structure
This is where the big and important volume of information is included.
This section is organised in 3 different interrelated hierarchies that
facilitate different points of view
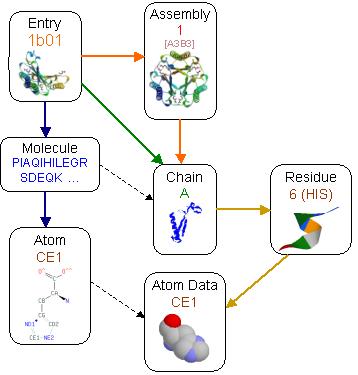 a) The sequence
point of view (denoted with blue arrows). The information in this hierarchy
is about the sequence and chemistry of the protein and does not relate
with the 3-D folding of this sequence. A molecule corresponds to the sequence
of a chain but it is possible to have more than one chain in the PDB asymmetric
unit that are slightly different foldings of the same molecule as these
were observed in the experiment. The atom is again the abstract notion
of a chemical atom that ignores alternative configurations or different
NMR models. These are useful in relationships where the actual coordinates
are not of interest, like the source organism of the molecule etc. a) The sequence
point of view (denoted with blue arrows). The information in this hierarchy
is about the sequence and chemistry of the protein and does not relate
with the 3-D folding of this sequence. A molecule corresponds to the sequence
of a chain but it is possible to have more than one chain in the PDB asymmetric
unit that are slightly different foldings of the same molecule as these
were observed in the experiment. The atom is again the abstract notion
of a chemical atom that ignores alternative configurations or different
NMR models. These are useful in relationships where the actual coordinates
are not of interest, like the source organism of the molecule etc.
b) The PDB asymmetric point of view (denoted with green and the green-orange
arrows). This is the view of the observed structure as is available in
the PDB entry. The asymmetric chains are also reused in assemblies
but are marked with a special non-symmetric-valid flag, that specifies
that are also valid regardless of the assembly where they belong. This
information is more useful when different chain structures are needed regardless
whether they are actually the same molecule and whether they have any interactions
between them.
c) The assembly point of view that corresponds to the actual quaternary
biological entity. This represents what should be considered as the actual
complete structure and is useful when the actual inter-chain and ligand interactions
are significant. For example the assembly in entry 1b01 above form a barrel
like sheet in the middle of the structure that includes strands from different
chains and becomes apparent after the assembly transformation of chains.
As an example the entry 1b01 has 5 chains in the asymmetric unit (A,B,C,D,E).
These chains form 3 assemblies, assembly 1 with chains (A,A1,A2,B,B1,B2),
assembly 2 with chains (C,C1,C2,D,D1,D2) and assembly 3 with chains (E,E1,E2,E3,E4,E5).
Chains A and B from assembly 1, C and D from assembly 2 and E from assembly
3, are also marked as non symmetric valid and they may be used to extract
the original PDB asymmetric unit.
Additionally all bound molecules and water groups are defined in separate
chains, named after and associated to the protein chains that have the
stronger interaction with. During the process of assembly formation,
bound molecules and waters may be replicated several times, as long as
they have some form of interaction with the assembly.
-
Secondary structure
This is a section of the database that keeps detailed information about
the secondary structure for common things like sheets and helices up to
more extended formations like bulges, hairpins and motifs. For each entry
there may be one or more sets of secondary structure information from different
sources. Since the secondary structure is not always available
in PDB entries and its source or accuracy is not consistent, the secondary
structure of all entries has been re-derived using directly the coordinates
of the structure as a source to DOSS, a secondary structure prediction program - based on
DSSP(W.Kabsch C. Sander(1983) Biopolymers 22:2577-2637) / Promotif [Gail
Hutchinson and Janet Thornton 1996], in order to provide an consistent platform for comparisons and
analysis of secondary structure. The starting point for deriving the secondary
structure information is not the PDB asymmetric unit, but the actual quaternary
structure (the assembly), in order to be able to identify secondary structure
elements related to more than 1 chain in the assembly. For example in entry
1b01 there is a barrel like sheet in the middle of the structure
that includes strands from 3-D transformed chains that originate from a
single chain of the asymmetric unit
-
Active sites
Information about the active sites of the macromolecule, and the way
that ligands and drugs bind to a protein. Again since the related information
sometimes available in the PDB entries is not consistent and trustworthy,
site information is calculated internally in PDBe
[Golovin, A., Dimitropoulos, D., Oldfield, T., Rachedi, A. and Henrick, K. (2005)
PROTEINS: Structure, Function, and Bioinformatics 58(1): 190-9.]
(http://www.ebi.ac.uk/msd-srv/msdsite/index.jsp).
The active sites of a protein chain are determined based on the contacts
of the chain with a ligand. There are many ways that contacts are defined
based on different types of bonds and interactions, that take into account
the distance and angles of the atoms, as well as other characteristics
of the ligands and residues like planes. An active site can be defined
not only for a particular atom, but also for a plane of a molecule.
-
External cross-references / Taxonomy
A lot of work has also been done to provide complete and consistent
cross-references with external database like Swiss-prot, SCOP, CATH, EC
Enzyme, Gene ontology, Medline and NCBI taxonomy databases
[Velankar, S., McNeil, P., Mittard-Runte, V., Suarez, A., Barrell, D., Apweiler, R. and Henrick.K. (2005) Nucleic Acids Res. 33 (Database Issue)].
The cross-references are established to the most suitable detailed level (for example on a residue
by residue basis for Swiss-prot, since the same chain may be referenced
by two different Swiss-prot entries) but are also often aggregated to facilitate
data analysis on a higher level. For more details on the broader context of this effort you may refer to
the eFamily web-site.
MSDSD frequently asked SQL
-
Get the reference info including atoms and bonds of a ligand (that could be used for example to dump out a chemical file)
-
Get the assemblies of an entry
Note: Assemblies with assembly serial 0 are not real biological assemblies. The serve for legacy purposes us placeholders for
data in the asymmetric unit (waters and ligands) that do not seem to be able to fit in a real assembly (problematic PDB data)
-
Get the chains in all assemblies of an entry (this is just for demonstration - not a actual useful query)
- Get the chains of an assembly
-
Get the chains of the asymmetric unit (original PDB file)
-
Get a single (ideally representative) chain per molecule
-
Get the residues of a chain
-
Get residues where the 3 letter code has been changed in the MSDSD (usually due to obsolete-superceded 3 letter codes
-
Get the residues and bound molecules that have not yet been associated with a reference ligand (to be cleaned)
-
Get the data to dump the "ATOM" lines of a PDB file for the original asymmetric unit file
-
Get the data to dump the PDB "ATOM" lines of an assembly
-
Get the data of a single (ideally the representative) NMR model for an NMR entry
-
Query for mySQL that pre-formats directly in the PDB format
(kindly contributed)
-
Get the helices of the original asymmetric unit of an entry
-
Get the actual and the observed aminoacid sequence string of the chains of an entry
Document mantained by: Gaurav Sahni
|