 |
PDBsum entry 5mw3
|
|

|
|
PDB id:
|
|
|
|
| Name: |
|
Transferase
|
|
|
Title:
|
|
Crystal structure of dot1l in complex with inhibitor cpd1 [n6-(2,6- dichlorophenyl)-n6-(pent-2-yn-1-yl)quinoline-4,6-diamine] and inhibitor cpd2 [(r)-1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-3- amine]
|
|
Structure:
|
|
Histone-lysine n-methyltransferase, h3 lysine-79 specific. Chain: a, b. Synonym: dot1-like protein,histone h3-k79 methyltransferase,h3-k79- hmtase,lysine n-methyltransferase 4. Engineered: yes
|
|
Source:
|
|
Homo sapiens. Human. Organism_taxid: 9606. Gene: dot1l, kiaa1814, kmt4. Expressed in: escherichia coli. Expression_system_taxid: 562
|
|
Resolution:
|
|
|
2.09Å
|
R-factor:
|
0.176
|
R-free:
|
0.193
|
|
|
Authors:
|
|
C.Be,E.Koch,C.Gaul,F.Stauffer,H.Moebitz,C.Scheufler
|
|
Key ref:
|
|
H.Möbitz
et al.
(2017).
Discovery of Potent, Selective, and Structurally Novel Dot1L Inhibitors by a Fragment Linking Approach.
Acs Med Chem Lett,
8,
338-343.
PubMed id:
|
|
|
Date:
|
|
|
18-Jan-17
|
Release date:
|
22-Mar-17
|
|
|
|
|
|
|
PROCHECK
|

|
|
|
|
|
Headers
|
|
|
|
References
|
|
|
|
|
|

|
|
Q8TEK3
(DOT1L_HUMAN) -
Histone-lysine N-methyltransferase, H3 lysine-79 specific from Homo sapiens
|
|
|
|
Seq:
Struc:
|
|
|
|
1537 a.a.
319 a.a.
|
|

|

|
|
|
|
|
|
|
|
|
|
|
|
Key: |
 |
PfamA domain |
|
|
 |
Secondary structure |
|
|
|
|
|
|
|
|
|
|
|
|
|
Enzyme class:
|
|
E.C.2.1.1.360
- [histone H3]-lysine(79) N-trimethyltransferase.
|
|
|
|
|
|
|
Reaction:
|
|
L-lysyl79-[histone H3] + 3 S-adenosyl-L-methionine = N6,N6,N6- trimethyl-L-lysyl79-[histone H3] + 3 S-adenosyl-L-homocysteine + 3 H+
|
|
|
|
|
|
L-lysyl(79)-[histone H3]
|
+
|
 3
×
S-adenosyl-L-methionine
3
×
S-adenosyl-L-methionine
|
=
|
N(6),N(6),N(6)- trimethyl-L-lysyl(79)-[histone H3]
|
+
|
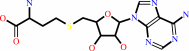 3
×
S-adenosyl-L-homocysteine
3
×
S-adenosyl-L-homocysteine
|
+
|
3
×
H(+)
Bound ligand (Het Group name = )
matches with 44.83% similarity
|
|
|
|
|
|
|
|
|
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
|
Acs Med Chem Lett
8:338-343
(2017)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
Discovery of Potent, Selective, and Structurally Novel Dot1L Inhibitors by a Fragment Linking Approach.
|
|
H.Möbitz,
R.Machauer,
P.Holzer,
A.Vaupel,
F.Stauffer,
C.Ragot,
G.Caravatti,
C.Scheufler,
C.Fernandez,
U.Hommel,
R.Tiedt,
K.S.Beyer,
C.Chen,
H.Zhu,
C.Gaul.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
Misdirected catalytic activity of histone methyltransferase Dot1L is believed to
be causative for a subset of highly aggressive acute leukemias. Targeting the
catalytic domain of Dot1L represents a potential therapeutic approach for these
leukemias. In the context of a comprehensive Dot1L hit finding strategy, a
knowledge-based virtual screen of the Dot1L SAM binding pocket led to the
discovery of 2, a non-nucleoside fragment mimicking key interactions of SAM
bound to Dot1L. Fragment linking of 2 and 3, an induced back pocket binder
identified in earlier studies, followed by careful ligand optimization led to
the identification of 7, a highly potent, selective and structurally novel Dot1L
inhibitor.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|


 Links
Links










