 |
PDBsum entry 2pyd

|
|
|
|
|
 |
Contents |
 |
|
|
|
|
|
|
|
|
|
|
|
|
* Residue conservation analysis
|
|
|
|
|
|
PDB id:
|
|
|
|
| Name: |
|
Transferase
|
|
|
Title:
|
|
The crystal structure of glycogen phosphorylase in complex with glucose at 100 k
|
|
Structure:
|
|
Glycogen phosphorylase, muscle form. Chain: a. Synonym: myophosphorylase. Ec: 2.4.1.1
|
|
Source:
|
|
Oryctolagus cuniculus. Rabbit. Organism_taxid: 9986
|
|
Resolution:
|
|
|
1.93Å
|
R-factor:
|
0.193
|
R-free:
|
0.235
|
|
|
Authors:
|
|
K.M.Alexacou,C.Tiraidis,S.E.Zographos,E.D.Chrysina,J.Hayes, N.G.Oikonomakos
|
Key ref:

|
|
K.M.Alexacou
et al.
(2008).
Crystallographic and computational studies on 4-phenyl-N-(beta-D-glucopyranosyl)-1H-1,2,3-triazole-1-acetamide, an inhibitor of glycogen phosphorylase: comparison with alpha-D-glucose, N-acetyl-beta-D-glucopyranosylamine and N-benzoyl-N'-beta-D-glucopyranosyl urea binding.
Proteins,
71,
1307-1323.
PubMed id:
DOI:

|
|
|
Date:
|
|
|
16-May-07
|
Release date:
|
01-Apr-08
|
|
|
|
|
|
|
PROCHECK
|

|
|
|
|
|
Headers
|
|
|
|
References
|
|
|
|
|
|

|
|
P00489
(PYGM_RABIT) -
Glycogen phosphorylase, muscle form from Oryctolagus cuniculus
|
|
|
|
Seq:
Struc:
|
|
|
|
843 a.a.
808 a.a.*
|
|

|

|

|
|
|
|
|
|
|
|
|
|
|
Key: |
 |
PfamA domain |
|
|
 |
Secondary structure |
|
 |
CATH domain |
|
|
*
PDB and UniProt seqs differ
at 1 residue position (black
cross)
|
|
|
|
|
|
|
|
|
|
|
|
|
Enzyme class:
|
|
E.C.2.4.1.1
- glycogen phosphorylase.
|
|
|
|
|
|
|
Pathway:
|
|
Glycogen
|
|
|
|
|
|
Reaction:
|
|
[(1->4)-alpha-D-glucosyl](n) + phosphate = [(1->4)-alpha-D-glucosyl](n-1) + alpha-D-glucose 1-phosphate
|
|
|
|
|
|
[(1->4)-alpha-D-glucosyl](n)
|
+
|
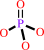 phosphate
phosphate
|
=
|
[(1->4)-alpha-D-glucosyl](n-1)
|
+
|
alpha-D-glucose 1-phosphate
Bound ligand (Het Group name = )
matches with 75.00% similarity
|
|
|
|
|
|
|
|
|
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
DOI no:
|
Proteins
71:1307-1323
(2008)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
Crystallographic and computational studies on 4-phenyl-N-(beta-D-glucopyranosyl)-1H-1,2,3-triazole-1-acetamide, an inhibitor of glycogen phosphorylase: comparison with alpha-D-glucose, N-acetyl-beta-D-glucopyranosylamine and N-benzoyl-N'-beta-D-glucopyranosyl urea binding.
|
|
K.M.Alexacou,
J.M.Hayes,
C.Tiraidis,
S.E.Zographos,
D.D.Leonidas,
E.D.Chrysina,
G.Archontis,
N.G.Oikonomakos,
J.V.Paul,
B.Varghese,
D.Loganathan.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
4-Phenyl-N-(beta-D-glucopyranosyl)-1H-1,2,3-triazole-1-acetamide
(glucosyltriazolylacetamide) has been studied in kinetic and crystallographic
experiments with glycogen phosphorylase b (GPb), in an effort to utilize its
potential as a lead for the design of potent antihyperglycaemic agents. Docking
and molecular dynamics (MD) calculations have been used to monitor more closely
the binding modes in operation and compare the results with experiment. Kinetic
experiments in the direction of glycogen synthesis showed that
glucosyltriazolylacetamide is a better inhibitor (K(i) = 0.18 mM) than the
parent compound alpha-D-glucose (K(i) = 1.7 mM) or beta-D-glucose (K(i) = 7.4
mM) but less potent inhibitor than the lead compound
N-acetyl-beta-D-glucopyranosylamine (K(i) = 32 microM). To elucidate the
molecular basis underlying the inhibition of the newly identified compound, we
determined the structure of GPb in complex with glucosyltriazolylacetamide at
100 K to 1.88 A resolution, and the structure of the compound in the free form.
Glucosyltriazolylacetamide is accommodated in the catalytic site of the enzyme
and the glucopyranose interacts in a manner similar to that observed in the
GPb-alpha-D-glucose complex, while the substituent group in the beta-position of
the C1 atom makes additional hydrogen bonding and van der Waals interactions to
the protein. A bifurcated donor type hydrogen bonding involving O3H, N3, and N4
is seen as an important structural motif strengthening the binding of
glucosyltriazolylacetamide with GP which necessitated change in the torsion
about C8-N2 bond by about 62 degrees going from its free to the complex form
with GPb. On binding to GP, glucosyltriazolylacetamide induces significant
conformational changes in the vicinity of this site. Specifically, the 280s loop
(residues 282-288) shifts 0.7 to 3.1 A (CA atoms) to accommodate
glucosyltriazolylacetamide. These conformational changes do not lead to
increased contacts between the inhibitor and the protein that would improve
ligand binding compared with the lead compound. In the molecular modeling
calculations, the GOLD docking runs with and without the crystallographic
ordered cavity waters using the GoldScore scoring function, and without cavity
waters using the ChemScore scoring function successfully reproduced the
crystallographic binding conformation. However, the GLIDE docking calculations
both with (GLIDE XP) and without (GLIDE SP and XP) the cavity water molecules
were, impressively, further able to accurately reproduce the finer details of
the GPb-glucosyltriazolylacetamide complex structure. The importance of cavity
waters in flexible receptor MD calculations compared to "rigid" (docking) is
analyzed and highlighted, while in the MD itself very little conformational
flexibility of the glucosyltriazolylacetamide ligand was observed over the time
scale of the simulations.
|
|
|
|
|
|
| |
Selected figure(s)
|
|
|
|
| |
|
|

|
|

|
|
Figure 6.
Figure 6. Stereo diagrams showing interactions between  -D-glucose
(a) and glucosyltriazolylacetamide (b) and protein in the
vicinity of the catalytic site. The hydrogen bond pattern
between ligands, protein residues, and water molecules (w) in
the catalytic site is represented by dotted lines. -D-glucose
(a) and glucosyltriazolylacetamide (b) and protein in the
vicinity of the catalytic site. The hydrogen bond pattern
between ligands, protein residues, and water molecules (w) in
the catalytic site is represented by dotted lines.
|
|
Figure 10.
Figure 10. The top-binding pose for the GLIDE XP docking which
was run including the cavity waters.
|
|
|
|
|
|
| |
The above figures are
reprinted
by permission from John Wiley & Sons, Inc.:
Proteins
(2008,
71,
1307-1323)
copyright 2008.
|
|
| |
Figures were
selected
by an automated process.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|
Literature references that cite this PDB file's key reference
|
|
|
| |
PubMed id
|
|
Reference
|
|
|
|
|
|
J.M.Hayes,
V.T.Skamnaki,
G.Archontis,
C.Lamprakis,
J.Sarrou,
N.Bischler,
A.L.Skaltsounis,
S.E.Zographos,
and
N.G.Oikonomakos
(2011).
Kinetics, in silico docking, molecular dynamics, and MM-GBSA binding studies on prototype indirubins, KT5720, and staurosporine as phosphorylase kinase ATP-binding site inhibitors: the role of water molecules examined.
|
| |
Proteins,
79,
703-719.
|
|
|
|
|
|
The most recent references are shown first.
Citation data come partly from CiteXplore and partly
from an automated harvesting procedure. Note that this is likely to be
only a partial list as not all journals are covered by
either method. However, we are continually building up the citation data
so more and more references will be included with time.
|
|


 Links
Links






