- Course overview
- Search within this course
- What is phylogenetics?
- Why is phylogenetics important?
- Major stages in phylogenetic analyses
- Phylogenetics resources at EMBL-EBI
- Discovering the phylogeny of all flowering plants
- Summary
- Quiz: test your knowledge
- Learn more
- Your feedback
- References
![]()
All materials are free cultural works licensed under a Creative Commons Attribution 4.0 International (CC BY 4.0) license, except where further licensing details are provided.
Branches
Branches show the path of transmission of genetic information from one generation to the next. Branch lengths indicate genetic change i.e. the longer the branch, the more genetic change (or divergence) has occurred. Typically we measure the extent of genetic change by estimating the average number of nucleotide or protein substitutions per site.
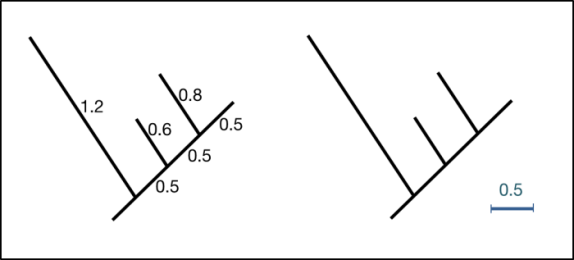
Informative branch lengths are typically drawn to scale and indicate the number of substitutions per site (Figure 7). Branch lengths are occasionally shown on the phylogeny (left), but it is far more common to see branch lengths represented by a scale bar (right). It can therefore be useful to keep a ruler close to hand for interpreting phylogenies that you see in the literature!
How do we estimate genetic change?
Estimating the extent of genetic change is not a trivial task. A naïve method is to align pairs of sequences, count up the number of differences and divide by the sequence length.

In the simple alignment above (Figure 8), we can see that there is one site that is different between the two sequences, and we could say that based upon this tiny sample there are 1/10 = 0.1 substitutions per site. However this assumes that we have observed every substitution that has happened, and therefore does not account any multiple substitutions that have occurred at any of the sites. We have also assumed that every substitution (e.g. from T>C, or A>G) is equally likely to have occurred, and we now know that this is unrealistic. To overcome these issues, it is now commonplace to use an evolutionary model to infer the genetic change that has occurred.
Beware of very long branches!
To get a value of one substitution per site using the simple method above would require the pair of sequences to be completely different to each other at all 10/10 sites. It is unlikely you would align such sequences since two random nucleotide sequences are likely to be 25% identical. So if you see figures in the literature with branches longer than ~3 substitutions per site then you might want to worry about the confidence we have in those estimates!