 |
PDBsum entry 2qe7

|
|
|
|
|
 |
Contents |
 |
|
|
|
|
|
|
|
|
|
|
|


 474 a.a.
474 a.a.
|
|
|
|
|
|
|
|
|
|
|


 461 a.a.
461 a.a.
|
|
|
|
|
|
|
|
|
|
|
 227 a.a.
227 a.a.
|
|
|
|
|
|
|
|
|
|
|
 135 a.a.
135 a.a.
|
|
|
|
|
|
|
* Residue conservation analysis
|
|
|
|
|
|
PDB id:
|
|
|
|
| Name: |
|
Hydrolase
|
|
|
Title:
|
|
Crystal structure of the f1-atpase from the thermoalkaliphilic bacterium bacillus sp. Ta2.A1
|
|
Structure:
|
|
Atp synthase subunit alpha. Chain: a, b, c. Engineered: yes. Atp synthase subunit beta. Chain: d, e, f. Engineered: yes. Atp synthase subunit gamma. Chain: g. Engineered: yes.
|
|
Source:
|
|
Bacillus sp.. Organism_taxid: 90973. Strain: ta2.A1. Gene: atpa. Expressed in: escherichia coli. Expression_system_taxid: 562. Gene: atpd. Gene: atpg. Gene: atpc.
|
|
Resolution:
|
|
|
3.06Å
|
R-factor:
|
0.252
|
R-free:
|
0.306
|
|
|
Authors:
|
|
A.Stocker,S.Keis,J.Vonck,G.M.Cook,P.Dimroth
|
Key ref:

|
|
A.Stocker
et al.
(2007).
The structural basis for unidirectional rotation of thermoalkaliphilic F1-ATPase.
Structure,
15,
904-914.
PubMed id:
DOI:

|
|
|
Date:
|
|
|
25-Jun-07
|
Release date:
|
21-Aug-07
|
|
|
|
|
|
|
PROCHECK
|

|
|
|
|
|
Headers
|
|
|
|
References
|
|
|
|
|
|

|
|
Q71CG5
(Q71CG5_9BACI) -
ATP synthase subunit alpha from Bacillus sp. TA2.A1
|
|
|
|
Seq:
Struc:
|
|
|
|
502 a.a.
474 a.a.
|
|

|

|

|
|
|
|
|
|
|
|
|
|
|
|

|
|
Q71CG3
(Q71CG3_9BACI) -
ATP synthase subunit beta from Bacillus sp. TA2.A1
|
|
|
|
Seq:
Struc:
|
|
|
|
462 a.a.
461 a.a.
|
|
|

|

|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Enzyme class 2:
|
|
Chains A, B, C, D, E, F:
E.C.7.1.2.2
- H(+)-transporting two-sector ATPase.
|
|
|
|
|
|
|
Reaction:
|
|
ATP + H2O + 4 H+(in) = ADP + phosphate + 5 H+(out)
|
|
|
|
|
|
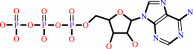 ATP
ATP
|
+
|
H2O
|
+
|
4
×
H(+)(in)
|
=
|
 ADP
ADP
|
+
|
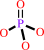 phosphate
phosphate
|
+
|
5
×
H(+)(out)
|
|
|
|
|
|
|
|
|
|
Enzyme class 3:
|
|
Chains G, H:
E.C.3.6.1.34
- Transferred entry: 7.1.2.2.
|
|
|
|
|
|
|
|
|
|
Note, where more than one E.C. class is given (as above), each may
correspond to a different protein domain or, in the case of polyprotein
precursors, to a different mature protein.
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
DOI no:
|
Structure
15:904-914
(2007)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
The structural basis for unidirectional rotation of thermoalkaliphilic F1-ATPase.
|
|
A.Stocker,
S.Keis,
J.Vonck,
G.M.Cook,
P.Dimroth.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
The ATP synthase of the thermoalkaliphilic Bacillus sp. TA2.A1 operates
exclusively in ATP synthesis direction. In the crystal structure of the
nucleotide-free alpha(3)beta(3)gamma epsilon subcomplex (TA2F(1)) at 3.1 A
resolution, all three beta subunits adopt the open beta(E) conformation. The
structure shows salt bridges between the helix-turn-helix motif of the
C-terminal domain of the beta(E) subunit (residues Asp372 and Asp375) and the
N-terminal helix of the gamma subunit (residues Arg9 and Arg10). These
electrostatic forces pull the gamma shaft out of the rotational center and
impede rotation through steric interference with the beta(E) subunit.
Replacement of Arg9 and Arg10 with glutamines eliminates the salt bridges and
results in an activation of ATP hydrolysis activity, suggesting that these salt
bridges prevent the native enzyme from rotating in ATP hydrolysis direction. A
similar bending of the gamma shaft as in the TA2F(1) structure was observed by
single-particle analysis of the TA2F(1)F(o) holoenzyme.
|
|
|
|
|
|
| |
Selected figure(s)
|
|
|
|
| |
|
|

|
|

|
|
Figure 1.
Figure 1. Stereo Drawings of the Three-Dimensional Structure
of the α[3]β[3]γ  epsilon
Subcomplex of F[1] from the Thermoalkaliphilic Bacillus sp.
TA2.A1 epsilon
Subcomplex of F[1] from the Thermoalkaliphilic Bacillus sp.
TA2.A1
(A) Side view of the complex with the pseudo
three-fold axis vertical. Three structural domains are visible
in each subunit of the α[3]β[3] hexamer. The β and α
subunits are shown in pink and pale yellow, respectively. The γ
subunit and the epsilon
subunit with the highest occupancy are shown in light blue and
lime, respectively.
(B) Top view of the complex viewed
toward the membrane; the pseudo three-fold axis points toward
the viewer.
(C) Bottom view of the complex viewed from the
membrane.
|
|
Figure 5.
Figure 5. Electron Microscopic Analysis of Native TA2F[1]F[o]
(A) Electron micrograph of TA2F[1]F[o] negatively stained
with uranyl acetate. The scale bar represents 50 nm.
(B)
Class averages after multivariate statistical analysis and
classification of 1940 particles. At least 20% of particles were
rejected based on poor resolution, as well as 20% from each
class. Classes 1–8 contain 197, 250, 197, 194, 184, 261, 141,
and 129 particles, respectively. Classes 1–4 show a view with
the central stalk attached asymmetrically on the left side of
the c ring; classes 5–7 show a more centrally connected stalk
and a much narrower F[o] domain. Class 8 contains some poorly
aligned particles, of which some have the F[1] domain facing
down. Each frame represents 382 Å.
(C) Corresponding
views of the TA2F[1] structure rotated approximately 90°
relative to each other. The images were created using UCSF
Chimera (Pettersen et al., 2004).
|
|
|
|
|
|
| |
The above figures are
reprinted
by permission from Cell Press:
Structure
(2007,
15,
904-914)
copyright 2007.
|
|
| |
Figures were
selected
by an automated process.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|
Literature references that cite this PDB file's key reference
|
|
|
| |
PubMed id
|
|
Reference
|
|
|
|
|
|
G.Cingolani,
and
T.M.Duncan
(2011).
Structure of the ATP synthase catalytic complex (F(1)) from Escherichia coli in an autoinhibited conformation.
|
| |
Nat Struct Mol Biol,
18,
701-707.
|
|
|
|
|
|
|
J.Vonck,
K.Y.Pisa,
N.Morgner,
B.Brutschy,
and
V.Müller
(2009).
Three-dimensional structure of A1A0 ATP synthase from the hyperthermophilic archaeon Pyrococcus furiosus by electron microscopy.
|
| |
J Biol Chem,
284,
10110-10119.
|
|
|
|
|
|
|
V.Kabaleeswaran,
H.Shen,
J.Symersky,
J.E.Walker,
A.G.Leslie,
and
D.M.Mueller
(2009).
Asymmetric structure of the yeast f1 ATPase in the absence of bound nucleotides.
|
| |
J Biol Chem,
284,
10546-10551.
|
|
|
PDB code:
|
|
|
|
|
|
|
|
C.von Ballmoos,
G.M.Cook,
and
P.Dimroth
(2008).
Unique rotary ATP synthase and its biological diversity.
|
| |
Annu Rev Biophys,
37,
43-64.
|
|
|
|
|
|
|
D.Pogoryelov,
Y.Nikolaev,
U.Schlattner,
K.Pervushin,
P.Dimroth,
and
T.Meier
(2008).
Probing the rotor subunit interface of the ATP synthase from Ilyobacter tartaricus.
|
| |
FEBS J,
275,
4850-4862.
|
|
|
|
|
|
|
H.Z.Mao,
C.G.Abraham,
A.M.Krishnakumar,
and
J.Weber
(2008).
A Functionally Important Hydrogen-bonding Network at the {beta}DP/{alpha}DP Interface of ATP Synthase.
|
| |
J Biol Chem,
283,
24781-24788.
|
|
|
|
|
|
|
J.J.García-Trejo,
and
E.Morales-Ríos
(2008).
Regulation of the F(1)F (0)-ATP Synthase Rotary Nanomotor in its Monomeric-Bacterial and Dimeric-Mitochondrial Forms.
|
| |
J Biol Phys,
34,
197-212.
|
|
|
|
|
|
|
M.A.Bianchet,
and
L.M.Amzel
(2007).
Making the right moves.
|
| |
Structure,
15,
885-886.
|
|
|
|
|
|
The most recent references are shown first.
Citation data come partly from CiteXplore and partly
from an automated harvesting procedure. Note that this is likely to be
only a partial list as not all journals are covered by
either method. However, we are continually building up the citation data
so more and more references will be included with time.
Where a reference describes a PDB structure, the PDB
code is
shown on the right.
|
| | |


 Links
Links











