 |
PDBsum entry 2gh7

|
|
|
|
|
|

|
|

|
|

|
|

|
|

|
|

|
|

|
|
|
 |

|

|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Biotin binding protein
|
PDB id
|
|
|
|
2gh7
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

|

|

|
|
|
|
|
|
References listed in PDB file
|
|
|
Key reference
|
|
|
Title
|
|
The high-Resolution structure of (+)-Epi-Biotin bound to streptavidin.
|
|
|
Authors
|
|
I.Le trong,
D.G.Aubert,
N.R.Thomas,
R.E.Stenkamp.
|
|
|
Ref.
|
|
Acta Crystallogr D Biol Crystallogr, 2006,
62,
576-581.
[DOI no: ]
|
|
|
PubMed id
|
|
|
|
|
|
Abstract
|
|
|
(+)-Epi-biotin differs from (+)-biotin in the configuration of the chiral center
at atom C2. This could lead to a difference in the mode of binding of
(+)-epi-biotin to streptavidin, a natural protein receptor for (+)-biotin.
Diffraction data were collected to a maximum of 0.85 Angstrom resolution for
structural analysis of the complex of streptavidin with a sample of
(+)-epi-biotin and refinement was carried out at both 1.0 and 0.85 Angstrom
resolution. The structure determination shows a superposition of two ligands in
the binding site, (+)-biotin and (+)-epi-biotin. The molecules overlap in the
model for the complex except for the position of S1 in the tetrahydrothiophene
ring. Differences in the conformation of the ring permits binding of each
molecule to streptavidin with little observable difference in the protein
structures at this high resolution.
|
|
|
|

|
|

|
|
Figure 2.
Figure 2 (a) Difference electron density for one binding site
after refinement including the biotin molecule in the model. (b)
Difference electron density after making the thermal parameter
of the S atom consistent with those of the neighboring atoms.
Negative densities are shown in red (-4  )
and positive in blue (4 )
and cyan (5 ). )
and positive in blue (4 )
and cyan (5 ).
|
|
Figure 5.
Figure 5 Difference electron-density map showing the positive
peaks associated with H atoms on a 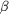 -strand
at the center of the molecule. Positive contours are shown at 3
(blue),
4 (purple)
and 5 (cyan);
negative contours are shown at -3 (red).
The |F[o]| - |F[c]| map was calculated using phases and |F[c]|
for the model just prior to the addition of H atoms at their
calculated positions. -strand
at the center of the molecule. Positive contours are shown at 3
(blue),
4 (purple)
and 5 (cyan);
negative contours are shown at -3 (red).
The |F[o]| - |F[c]| map was calculated using phases and |F[c]|
for the model just prior to the addition of H atoms at their
calculated positions.
|
|
|
|
|
|
The above figures are
reprinted
by permission from the IUCr:
Acta Crystallogr D Biol Crystallogr
(2006,
62,
576-581)
copyright 2006.
|
|
|
|
|
|
|


