 |
PDBsum entry 7knh

|
|
|
|
|
|

|

|

|

|
|

|
|

|
|

|
|

|
|

|
|
|
 |

|

|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Hydrolase/viral protein
|
PDB id
|
|
|
|
7knh
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

|

|

|
|
|
|
|
|
|
PDB id:
|
|
|
|
| Name: |
|
Hydrolase/viral protein
|
|
|
Title:
|
|
Cryo-em structure of double ace2-bound sars-cov-2 trimer spike at ph 5.5
|
|
Structure:
|
|
Spike glycoprotein. Chain: c, a, b. Synonym: s glycoprotein,e2,peplomer protein,sars-cov-2 spike glycoprotein. Engineered: yes. Angiotensin-converting enzyme 2. Chain: e, d. Synonym: ace-related carboxypeptidase,angiotensin-converting enzyme homolog,aceh,metalloprotease mprot15.
|
|
Source:
|
|
Severe acute respiratory syndrome coronavirus 2. 2019-ncov. Organism_taxid: 2697049. Gene: s, 2. Expressed in: homo sapiens. Expression_system_taxid: 9606. Homo sapiens. Human.
|
|
Authors:
|
|
J.Gorman,M.Rapp,P.D.Kwong,L.Shapiro
|
|
Key ref:
|
|
T.Zhou
et al.
(2020).
Cryo-EM Structures of SARS-CoV-2 Spike without and with ACE2 Reveal a pH-Dependent Switch to Mediate Endosomal Positioning of Receptor-Binding Domains.
Cell Host Microbe,
28,
867.
PubMed id:
|
|
|
Date:
|
|
|
04-Nov-20
|
Release date:
|
16-Dec-20
|
|
|
|
|
|
|
PROCHECK
|

|
|
|
|
|
Headers
|
|
|
|
References
|
|
|
|
|
|
|
|
|
|
|
|
Enzyme class 1:
|
|
Chains E, D:
E.C.3.4.17.-
- ?????
|
|
|
|
|
|
|
Enzyme class 2:
|
|
Chains E, D:
E.C.3.4.17.23
- angiotensin-converting enzyme 2.
|
|
|
|
|
|
|
Reaction:
|
|
angiotensin II + H2O = angiotensin-1-7 + L-phenylalanine
|
|
|
|
|
|
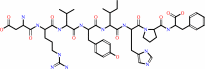 angiotensin II
angiotensin II
|
+
|
H2O
|
=
|
angiotensin-(1-7)
|
+
|
L-phenylalanine
Bound ligand (Het Group name = )
matches with 44.44% similarity
|
|
|
|
|
|
|
|
|
|
|
|
|
Note, where more than one E.C. class is given (as above), each may
correspond to a different protein domain or, in the case of polyprotein
precursors, to a different mature protein.
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
|
Cell Host Microbe
28:867
(2020)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
Cryo-EM Structures of SARS-CoV-2 Spike without and with ACE2 Reveal a pH-Dependent Switch to Mediate Endosomal Positioning of Receptor-Binding Domains.
|
|
T.Zhou,
Y.Tsybovsky,
J.Gorman,
M.Rapp,
G.Cerutti,
G.Y.Chuang,
P.S.Katsamba,
J.M.Sampson,
A.Schön,
J.Bimela,
J.C.Boyington,
A.Nazzari,
A.S.Olia,
W.Shi,
M.Sastry,
T.Stephens,
J.Stuckey,
I.T.Teng,
P.Wang,
S.Wang,
B.Zhang,
R.A.Friesner,
D.D.Ho,
J.R.Mascola,
L.Shapiro,
P.D.Kwong.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
The SARS-CoV-2 spike employs mobile receptor-binding domains (RBDs) to engage
the human ACE2 receptor and to facilitate virus entry, which can occur through
low-pH-endosomal pathways. To understand how ACE2 binding and low pH affect
spike conformation, we determined cryo-electron microscopy structures-at
serological and endosomal pH-delineating spike recognition of up to three ACE2
molecules. RBDs freely adopted "up" conformations required for ACE2
interaction, primarily through RBD movement combined with smaller alterations in
neighboring domains. In the absence of ACE2, single-RBD-up conformations
dominated at pH 5.5, resolving into a solitary all-down conformation at lower
pH. Notably, a pH-dependent refolding region (residues 824-858) at the
spike-interdomain interface displayed dramatic structural rearrangements and
mediated RBD positioning through coordinated movements of the entire trimer
apex. These structures provide a foundation for understanding prefusion-spike
mechanics governing endosomal entry; we suggest that the low pH all-down
conformation potentially facilitates immune evasion from RBD-up binding antibody.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|
|


 Links
Links


















