 |
PDBsum entry 5ngr
|
|

|
|
PDB id:
|
|
|
|
| Name: |
|
Hydrolase
|
|
|
Title:
|
|
Crystal structure of human mth1 in complex with fragment inhibitor 8- (methylsulfanyl)-7h-purin-6-amine
|
|
Structure:
|
|
7,8-dihydro-8-oxoguanine triphosphatase. Chain: a, b. Fragment: unp residues 42-197. Synonym: 2-hydroxy-datp diphosphatase,8-oxo-dgtpase,nucleoside diphosphate-linked moiety x motif 1,nudix motif 1. Engineered: yes
|
|
Source:
|
|
Homo sapiens. Human. Organism_taxid: 9606. Gene: nudt1, mth1. Expressed in: escherichia coli bl21(de3). Expression_system_taxid: 469008
|
|
Resolution:
|
|
|
2.20Å
|
R-factor:
|
0.213
|
R-free:
|
0.254
|
|
|
Authors:
|
|
R.Gustafsson,A.Rudling,I.Almlof,E.Homan,M.Scobie,U.Warpman Berglund, T.Helleday,J.Carlsson,P.Stenmark
|
|
Key ref:
|
|
A.Rudling
et al.
(2017).
Fragment-Based Discovery and Optimization of Enzyme Inhibitors by Docking of Commercial Chemical Space.
J Med Chem,
60,
8160-8169.
PubMed id:
DOI:
|
|
|
Date:
|
|
|
20-Mar-17
|
Release date:
|
04-Oct-17
|
|
|
|
|
|
|
PROCHECK
|

|
|
|
|
|
Headers
|
|
|
|
References
|
|
|
|
|
|

|
|
P36639
(8ODP_HUMAN) -
Oxidized purine nucleoside triphosphate hydrolase from Homo sapiens
|
|
|
|
Seq:
Struc:
|
|
|
|
156 a.a.
154 a.a.
|
|

|

|
|
|
|
|
|
|
|
|
|
|
|
Key: |
 |
PfamA domain |
|
|
 |
Secondary structure |
|
|
|
|
|
|
|
|
|
|
|
|
|
Enzyme class 2:
|
|
E.C.3.6.1.-
- ?????
|
|
|
|
|
|
|
Enzyme class 3:
|
|
E.C.3.6.1.56
- 2-hydroxy-dATP diphosphatase.
|
|
|
|
|
|
|
Reaction:
|
|
2-oxo-dATP + H2O = 2-oxo-dAMP + diphosphate + H+
|
|
|
|
|
|
2-oxo-dATP
|
+
|
H2O
|
=
|
2-oxo-dAMP
|
+
|
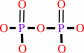 diphosphate
diphosphate
|
+
|
H(+)
|
|
|
|
|
|
|
|
|
|
|
|
|
Note, where more than one E.C. class is given (as above), each may
correspond to a different protein domain or, in the case of polyprotein
precursors, to a different mature protein.
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
DOI no:
|
J Med Chem
60:8160-8169
(2017)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
Fragment-Based Discovery and Optimization of Enzyme Inhibitors by Docking of Commercial Chemical Space.
|
|
A.Rudling,
R.Gustafsson,
I.Almlöf,
E.Homan,
M.Scobie,
U.Warpman Berglund,
T.Helleday,
P.Stenmark,
J.Carlsson.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
Fragment-based lead discovery has emerged as a leading drug development strategy
for novel therapeutic targets. Although fragment-based drug discovery benefits
immensely from access to atomic-resolution information, structure-based virtual
screening has rarely been used to drive fragment discovery and optimization.
Here, molecular docking of 0.3 million fragments to a crystal structure of
cancer target MTH1 was performed. Twenty-two predicted fragment ligands, for
which analogs could be acquired commercially, were experimentally evaluated.
Five fragments inhibited MTH1 with IC50values ranging from 6 to 79
μM. Structure-based optimization guided by predicted binding modes and analogs
from commercial chemical libraries yielded nanomolar inhibitors. Subsequently
solved crystal structures confirmed binding modes predicted by docking for three
scaffolds. Structure-guided exploration of commercial chemical space using
molecular docking gives access to fragment libraries that are several orders of
magnitude larger than those screened experimentally and can enable efficient
optimization of hits to potent leads.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|


 Links
Links




