 |
PDBsum entry 3a8x

|
|
|
|
|
 |
Contents |
 |
|
|
|
|
|
|
|
|
|
|
|
|
* Residue conservation analysis
|
|
|
|
|
|
|
|
|
|
|
Enzyme class:
|
|
E.C.2.7.11.13
- protein kinase C.
|
|
|
|
|
|
|
Reaction:
|
|
|
1.
|
L-seryl-[protein] + ATP = O-phospho-L-seryl-[protein] + ADP + H+
|
|
2.
|
L-threonyl-[protein] + ATP = O-phospho-L-threonyl-[protein] + ADP + H+
|
|
|
|
|
|
|
L-seryl-[protein]
|
+
|
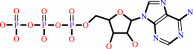 ATP
ATP
|
=
|
O-phospho-L-seryl-[protein]
|
+
|
 ADP
ADP
|
+
|
H(+)
|
|
|
|
|
|
|
L-threonyl-[protein]
|
+
|
ATP
|
=
|
O-phospho-L-threonyl-[protein]
|
+
|
ADP
|
+
|
H(+)
|
|
|
|
|
|
|
|
|
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
|
Acta Crystallogr D Biol Crystallogr
66:577-583
(2010)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
Structures of the PKC-iota kinase domain in its ATP-bound and apo forms reveal defined structures of residues 533-551 in the C-terminal tail and their roles in ATP binding.
|
|
T.Takimura,
K.Kamata,
K.Fukasawa,
H.Ohsawa,
H.Komatani,
T.Yoshizumi,
I.Takahashi,
H.Kotani,
Y.Iwasawa.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
Protein kinase C (PKC) plays an essential role in a wide range of cellular
functions. Although crystal structures of the PKC-theta, PKC-iota and PKC-betaII
kinase domains have previously been determined in complexes with small-molecule
inhibitors, no structure of a PKC-substrate complex has been determined. In the
previously determined PKC-iota complex, residues 533-551 in the C-terminal tail
were disordered. In the present study, crystal structures of the PKC-iota kinase
domain in its ATP-bound and apo forms were determined at 2.1 and 2.0 A
resolution, respectively. In the ATP complex, the electron density of all of the
C-terminal tail residues was well defined. In the structure, the side chain of
Phe543 protrudes into the ATP-binding pocket to make van der Waals interactions
with the adenine moiety of ATP; this is also observed in other AGC kinase
structures such as binary and ternary substrate complexes of PKA and AKT. In
addition to this interaction, the newly defined residues around the turn motif
make multiple hydrogen bonds to glycine-rich-loop residues. These interactions
reduce the flexibility of the glycine-rich loop, which is organized for ATP
binding, and the resulting structure promotes an ATP conformation that is
suitable for the subsequent phosphoryl transfer. In the case of the apo form,
the structure and interaction mode of the C-terminal tail of PKC-iota are
essentially identical to those of the ATP complex. These results indicate that
the protein structure is pre-organized before substrate binding to PKC-iota,
which is different from the case of the prototypical AGC-branch kinase PKA.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|


 Links
Links












