 |
PDBsum entry 2pnr

|
|
|
|
|
 |
Contents |
 |
|
|
|
|
|
|
|
|
|
|
|

 374 a.a.
374 a.a.
|
|
|
|
|
|
|
|
|
|
|

 341 a.a.
341 a.a.
|
|
|
|
|
|
|
|
|
|
|

 79 a.a.
79 a.a.
|
|
|
|
|
|
|
|
|
|
|
* Residue conservation analysis
|
|
|
|
|
|
PDB id:
|
|
|
|
| Name: |
|
Transferase
|
|
|
Title:
|
|
Crystal structure of the asymmetric pdk3-l2 complex
|
|
Structure:
|
|
[Pyruvate dehydrogenase [lipoamide]] kinase isozyme 3. Chain: a, b, e, f. Synonym: pyruvate dehydrogenase kinase isoform 3. Engineered: yes. Dihydrolipoyllysine-residue acetyltransferase component of pyruvate dehydrogenase complex. Chain: c, g. Synonym: pyruvate dehydrogenase complex e2 subunit, pdce2, e2, dihydrolipoamide s-acetyltransferase component of pyruvate
|
|
Source:
|
|
Homo sapiens. Human. Organism_taxid: 9606. Gene: pdk3. Expressed in: escherichia coli bl21. Expression_system_taxid: 511693. Gene: dlat, dlta.
|
|
Resolution:
|
|
|
2.50Å
|
R-factor:
|
0.176
|
R-free:
|
0.229
|
|
|
Authors:
|
|
D.G.Vassylyev,C.N.Steussy,Y.Devedjiev
|
Key ref:

|
|
Y.Devedjiev
et al.
(2007).
Crystal structure of an asymmetric complex of pyruvate dehydrogenase kinase 3 with lipoyl domain 2 and its biological implications.
J Mol Biol,
370,
407-416.
PubMed id:
DOI:

|
|
|
Date:
|
|
|
25-Apr-07
|
Release date:
|
21-Aug-07
|
|
|
|
|
|
|
PROCHECK
|

|
|
|
|
|
Headers
|
|
|
|
References
|
|
|
|
|
|

|
|
Q15120
(PDK3_HUMAN) -
[Pyruvate dehydrogenase (acetyl-transferring)] kinase isozyme 3, mitochondrial from Homo sapiens
|
|
|
|
Seq:
Struc:
|
|
|
|
406 a.a.
374 a.a.
|
|

|

|

|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Enzyme class 2:
|
|
Chains A, B, E, F:
E.C.2.7.11.2
- [pyruvate dehydrogenase (acetyl-transferring)] kinase.
|
|
|
|
|
|
|
Reaction:
|
|
L-seryl-[pyruvate dehydrogenase E1 alpha subunit] + ATP = O-phospho-L- seryl-[pyruvate dehydrogenase E1 alpha subunit] + ADP + H+
|
|
|
|
|
|
L-seryl-[pyruvate dehydrogenase E1 alpha subunit]
|
+
|
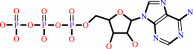 ATP
ATP
|
=
|
O-phospho-L- seryl-[pyruvate dehydrogenase E1 alpha subunit]
|
+
|
 ADP
ADP
|
+
|
H(+)
|
|
|
|
|
|
|
|
|
|
Enzyme class 3:
|
|
Chains C, G:
E.C.2.3.1.12
- dihydrolipoyllysine-residue acetyltransferase.
|
|
|
|
|
|
|
Pathway:
|
|
|
|
|
|
|
|
Reaction:
|
|
N6-[(R)-dihydrolipoyl]-L-lysyl-[protein] + acetyl-CoA = N6-[(R)-S(8)- acetyldihydrolipoyl]-L-lysyl-[protein] + CoA
|
|
|
|
|
|
N(6)-[(R)-dihydrolipoyl]-L-lysyl-[protein]
|
+
|
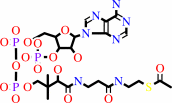 acetyl-CoA
acetyl-CoA
|
=
|
N(6)-[(R)-S(8)- acetyldihydrolipoyl]-L-lysyl-[protein]
|
+
|
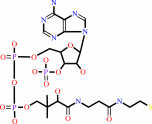 CoA
CoA
|
|
|
|
|
|
|
|
|
|
|
|
|
Note, where more than one E.C. class is given (as above), each may
correspond to a different protein domain or, in the case of polyprotein
precursors, to a different mature protein.
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
DOI no:
|
J Mol Biol
370:407-416
(2007)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
Crystal structure of an asymmetric complex of pyruvate dehydrogenase kinase 3 with lipoyl domain 2 and its biological implications.
|
|
Y.Devedjiev,
C.N.Steussy,
D.G.Vassylyev.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
A homodimer of pyruvate dehydrogenase kinase (PDHK) is an integral part of
pyruvate dehydrogenase complex (PDC) to which it is anchored primarily through
the inner lipoyl-bearing domains (L2) of transacetylase component. The catalytic
cycle of PDHK and its translocation over the PDC surface is thought to be
mediated by the "symmetric" and "asymmetric" modes, in which
the PDHK dimer binds to two and one L2-domain(s), respectively. Whereas the
structure of the symmetric PDHK/L2 complex was reported, the structural
organization and functional role of the asymmetric complex remain obscure. Here,
we report the crystal structure of the asymmetric PDHK3/L2 complex that reveals
several functionally important features absent from the previous structures.
First, the PDHK3 subunits have distinct conformations: one subunit exhibits
"open" and the other "closed" configuration of the putative
substrate-binding cleft. Second, access to the closed cleft is additionally
restricted by local unwinding of the adjacent alpha-helix. Modeling indicates
that the target peptide might gain access to the PDHK active center through the
open but not through the closed cleft. Third, the ATP-binding loop in one PDHK3
subunit adopts an open conformation, implying that the nucleotide loading into
the active site is mediated by the inactive "pre-insertion" binding
mode. Altogether our data suggest that the asymmetric complex represents a
physiological state in which binding of a single L2-domain activates one of the
PDHK protomers while inactivating another. Thus, the L2-domains likely act not
only as the structural anchors but also modulate the catalytic cycle of PDHK.
|
|
|
|
|
|
| |
Selected figure(s)
|
|
|
|
| |
|
|

|
|

|
|
Figure 2.
Figure 2. The gate helices in the L2-free and L2-bound
subunits. (a) and (b) The L2-free open (a) and L2-bound (b)
configurations of the SC (marked by yellow hydrophobic residues)
correspond to the uniform (magenta (a)) and distorted (orange
(b)) conformations of the gate helix. The remainder of the
subunits is shown in gray. The PDHK active site is marked by the
modeled (PDB ID 1Y8P) ATP molecule. (c) and (d) Superposition of
the L2-free (magenta (c)) and L2-bound (orange (d)) PDHK gate
helices with the RNA polymerase bridge helix (white) observed in
the uniform (c) and locally distorted (d) conformations. (e)
Stereo view of the L2-bound subunit showing stabilization of the
partially unwound gate helix. The hydrogen bonds are shown by
magenta broken lines. The active site is marked by ATP as in (a)
and (b). The views in (a), (b), and (e) are the same as in
Figure 1(b).
|
|
Figure 3.
Figure 3. Modeling of the E1/PDHK complex. (a) The overall
stereo view of the E1 α-subunit with the modeled α-helical
substrate peptide (yellow) docked into the SC of the L2-free
PDHK subunit. The E1 β-chain and PDHK L2-bound subunit are
located far from the modeled interface and are omitted from the
model for clarity. (b) The close-up stereo view of the interface
between the E1 substrate α-helix (yellow) and the SC.
Hydrophobic side-chains of the substrate are shown in yellow,
the phosphorylation site (Ser264) that is modeled in close
proximity (  vert,
similar 3.3 Å) to the ATP (green) γ-phosphate is colored
in orange. The PDHK backbone and hydrophobic side-chains are
shown in the domain-dependent colors corresponding to those in
(a). The view is roughly the same as in Figure 1(b). (c)
Schematic drawing of the putative van der Waals interactions
(broken lines) that may be formed between the E1 substrate
α-helix and the PDHK hydrophobic residues in the SC. The color
scheme is the same as in (a) and (b). vert,
similar 3.3 Å) to the ATP (green) γ-phosphate is colored
in orange. The PDHK backbone and hydrophobic side-chains are
shown in the domain-dependent colors corresponding to those in
(a). The view is roughly the same as in Figure 1(b). (c)
Schematic drawing of the putative van der Waals interactions
(broken lines) that may be formed between the E1 substrate
α-helix and the PDHK hydrophobic residues in the SC. The color
scheme is the same as in (a) and (b).
|
|
|
|
|
|
| |
The above figures are
reprinted
by permission from Elsevier:
J Mol Biol
(2007,
370,
407-416)
copyright 2007.
|
|
| |
Figures were
selected
by the author.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|
Literature references that cite this PDB file's key reference
|
|
|
| |
PubMed id
|
|
Reference
|
|
|
|
|
|
M.Kato,
J.Li,
J.L.Chuang,
and
D.T.Chuang
(2007).
Distinct structural mechanisms for inhibition of pyruvate dehydrogenase kinase isoforms by AZD7545, dichloroacetate, and radicicol.
|
| |
Structure,
15,
992.
|
|
|
PDB codes:
|
|
|
|
|
|
|
The most recent references are shown first.
Citation data come partly from CiteXplore and partly
from an automated harvesting procedure. Note that this is likely to be
only a partial list as not all journals are covered by
either method. However, we are continually building up the citation data
so more and more references will be included with time.
Where a reference describes a PDB structure, the PDB
codes are
shown on the right.
|
|
| |


 Links
Links














