 |
PDBsum entry 2plm

|
|
|
|
|
|

|

|

|

|
|

|
|

|
|

|
|

|
|
|
 |

|

|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Unknown function
|
PDB id
|
|
|
|
2plm
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

|

|

|
|
|
|
|
|
|
|
 |
Contents |
 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
* Residue conservation analysis
|
|
|
|
|
|
|
|
|
|
|
Enzyme class 1:
|
|
E.C.3.5.4.28
- S-adenosylhomocysteine deaminase.
|
|
|
|
|
|
|
Reaction:
|
|
S-adenosyl-L-homocysteine + H2O + H+ = S-inosyl-L-homocysteine + NH4+
|
|
|
|
|
|
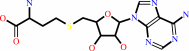 S-adenosyl-L-homocysteine
S-adenosyl-L-homocysteine
|
+
|
H2O
|
+
|
H(+)
|
=
|
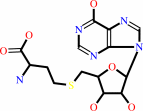 S-inosyl-L-homocysteine
S-inosyl-L-homocysteine
|
+
|
NH4(+)
Bound ligand (Het Group name = )
corresponds exactly
|
|
|
|
|
|
|
|
|
|
Enzyme class 2:
|
|
E.C.3.5.4.31
- S-methyl-5'-thioadenosine deaminase.
|
|
|
|
|
|
|
Reaction:
|
|
S-methyl-5'-thioadenosine + H2O + H+ = S-methyl-5'-thioinosine + NH4+
|
|
|
|
|
|
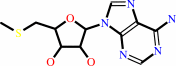 S-methyl-5'-thioadenosine
S-methyl-5'-thioadenosine
|
+
|
H2O
|
+
|
H(+)
Bound ligand (Het Group name = )
matches with 70.37% similarity
|
=
|
S-methyl-5'-thioinosine
|
+
|
NH4(+)
|
|
|
|
|
|
|
|
|
|
|
|
|
Note, where more than one E.C. class is given (as above), each may
correspond to a different protein domain or, in the case of polyprotein
precursors, to a different mature protein.
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
DOI no:
|
Nature
448:775-779
(2007)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
Structure-based activity prediction for an enzyme of unknown function.
|
|
J.C.Hermann,
R.Marti-Arbona,
A.A.Fedorov,
E.Fedorov,
S.C.Almo,
B.K.Shoichet,
F.M.Raushel.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
With many genomes sequenced, a pressing challenge in biology is predicting the
function of the proteins that the genes encode. When proteins are unrelated to
others of known activity, bioinformatics inference for function becomes
problematic. It would thus be useful to interrogate protein structures for
function directly. Here, we predict the function of an enzyme of unknown
activity, Tm0936 from Thermotoga maritima, by docking high-energy intermediate
forms of thousands of candidate metabolites. The docking hit list was dominated
by adenine analogues, which appeared to undergo C6-deamination. Four of these,
including 5-methylthioadenosine and S-adenosylhomocysteine (SAH), were tested as
substrates, and three had substantial catalytic rate constants (10(5) M(-1
)s(-1)). The X-ray crystal structure of the complex between Tm0936 and the
product resulting from the deamination of SAH, S-inosylhomocysteine, was
determined, and it corresponded closely to the predicted structure. The
deaminated products can be further metabolized by T. maritima in a previously
uncharacterized SAH degradation pathway. Structure-based docking with
high-energy forms of potential substrates may be a useful tool to annotate
enzymes for function.
|
|
|
|
|
|
| |
Selected figure(s)
|
|
|
|
| |
|
|

|
|

|
|
Figure 1.
Figure 1: Sample transformations of metabolites from their
ground state structure into the high-energy intermediate forms
that were used for docking. Transformations were computed
according to the conserved reaction mechanism of
amidohydrolases, a nucleophilic attack of a hydroxide at an
electrophilic centre atom. Every transformable functional group
for each molecule was processed independently. If the
high-energy structure was chiral, all stereoisomers were
calculated. Reactions catalysed by the amidohydrolases cytosine
deaminase (CDA), adenosine deaminase (ADA), dihydroorotase
(DHO), D-hydantoinase (HYD), isoaspartyl-d-dipeptidase (IAD),
N-acetyl-D-glucosamine-6-phosphate deacetylase (NaGA) and
phosphotriesterase (PTE) are shown.
|
|
Figure 3.
Figure 3: Comparing the docking prediction and the
crystallographic result.
Figure 3 : Comparing the docking
prediction and the crystallographic result.
Superposition of the crystal structure of Tm0936 in complex
with SIH (red) and the docking predicted structure of the
high-energy intermediate of SAH (carbons in green). Enzyme
carbons are coloured light blue, SAH and enzyme oxygen atoms are
coloured red, nitrogens blue and sulphurs orange. The purple
sphere represents the divalent metal ion. An F[O] – F[C] omit
electron density map for SIH is shown, contoured at 4.1  .
The structure was determined at 2.1 Å resolution. .
The structure was determined at 2.1 Å resolution.
|
|
|
|
|
|
| |
The above figures are
reprinted
by permission from Macmillan Publishers Ltd:
Nature
(2007,
448,
775-779)
copyright 2007.
|
|
| |
Figures were
selected
by an automated process.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|
Literature references that cite this PDB file's key reference
|
|
|
| |
PubMed id
|
|
Reference
|
|
|
|
|
|
Y.Qu,
and
J.C.Spain
(2011).
Catabolic pathway for 2-nitroimidazole involves a novel nitrohydrolase that also confers drug resistance.
|
| |
Environ Microbiol,
13,
1010-1017.
|
|
|
|
|
|
|
A.D.Hanson,
A.Pribat,
J.C.Waller,
and
V.de Crécy-Lagard
(2010).
'Unknown' proteins and 'orphan' enzymes: the missing half of the engineering parts list--and how to find it.
|
| |
Biochem J,
425,
1.
|
|
|
|
|
|
|
C.Kalyanaraman,
and
M.P.Jacobson
(2010).
Studying enzyme-substrate specificity in silico: a case study of the Escherichia coli glycolysis pathway.
|
| |
Biochemistry,
49,
4003-4005.
|
|
|
|
|
|
|
D.E.Almonacid,
E.R.Yera,
J.B.Mitchell,
and
P.C.Babbitt
(2010).
Quantitative comparison of catalytic mechanisms and overall reactions in convergently evolved enzymes: implications for classification of enzyme function.
|
| |
PLoS Comput Biol,
6,
e1000700.
|
|
|
|
|
|
|
J.A.Cummings,
T.T.Nguyen,
A.A.Fedorov,
P.Kolb,
C.Xu,
E.V.Fedorov,
B.K.Shoichet,
D.P.Barondeau,
S.C.Almo,
and
F.M.Raushel
(2010).
Structure, mechanism, and substrate profile for Sco3058: the closest bacterial homologue to human renal dipeptidase .
|
| |
Biochemistry,
49,
611-622.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
L.P.de Carvalho,
H.Zhao,
C.E.Dickinson,
N.M.Arango,
C.D.Lima,
S.M.Fischer,
O.Ouerfelli,
C.Nathan,
and
K.Y.Rhee
(2010).
Activity-based metabolomic profiling of enzymatic function: identification of Rv1248c as a mycobacterial 2-hydroxy-3-oxoadipate synthase.
|
| |
Chem Biol,
17,
323-332.
|
|
|
|
|
|
|
M.Bokhove,
H.Yoshida,
C.M.Hensgens,
J.M.van der Laan,
J.D.Sutherland,
and
B.W.Dijkstra
(2010).
Structures of an isopenicillin N converting Ntn-hydrolase reveal different catalytic roles for the active site residues of precursor and mature enzyme.
|
| |
Structure,
18,
301-308.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
M.Bucci,
C.Goodman,
and
T.L.Sheppard
(2010).
A decade of chemical biology.
|
| |
Nat Chem Biol,
6,
847-854.
|
|
|
|
|
|
|
M.J.Keiser,
J.J.Irwin,
and
B.K.Shoichet
(2010).
The chemical basis of pharmacology.
|
| |
Biochemistry,
49,
10267-10276.
|
|
|
|
|
|
|
M.Moll,
D.H.Bryant,
and
L.E.Kavraki
(2010).
The LabelHash algorithm for substructure matching.
|
| |
BMC Bioinformatics,
11,
555.
|
|
|
|
|
|
|
R.G.Coleman,
and
K.A.Sharp
(2010).
Protein pockets: inventory, shape, and comparison.
|
| |
J Chem Inf Model,
50,
589-603.
|
|
|
|
|
|
|
R.S.Hall,
A.A.Fedorov,
R.Marti-Arbona,
E.V.Fedorov,
P.Kolb,
J.M.Sauder,
S.K.Burley,
B.K.Shoichet,
S.C.Almo,
and
F.M.Raushel
(2010).
The hunt for 8-oxoguanine deaminase.
|
| |
J Am Chem Soc,
132,
1762-1763.
|
|
|
PDB code:
|
|
|
|
|
|
|
|
S.Mondal,
C.Nagao,
and
K.Mizuguchi
(2010).
Detecting subtle functional differences in ketopantoate reductase and related enzymes using a rule-based approach with sequence-structure homology recognition scores.
|
| |
Protein Eng Des Sel,
23,
859-869.
|
|
|
|
|
|
|
V.Vacic,
L.M.Iakoucheva,
S.Lonardi,
and
P.Radivojac
(2010).
Graphlet kernels for prediction of functional residues in protein structures.
|
| |
J Comput Biol,
17,
55-72.
|
|
|
|
|
|
|
A.J.Smith,
Y.Li,
and
K.N.Houk
(2009).
Quantum mechanics/molecular mechanics investigation of the mechanism of phosphate transfer in human uridine-cytidine kinase 2.
|
| |
Org Biomol Chem,
7,
2716-2724.
|
|
|
|
|
|
|
D.F.Xiang,
C.Xu,
D.Kumaran,
A.C.Brown,
J.M.Sauder,
S.K.Burley,
S.Swaminathan,
and
F.M.Raushel
(2009).
Functional annotation of two new carboxypeptidases from the amidohydrolase superfamily of enzymes.
|
| |
Biochemistry,
48,
4567-4576.
|
|
|
|
|
|
|
D.F.Xiang,
Y.Patskovsky,
C.Xu,
A.J.Meyer,
J.M.Sauder,
S.K.Burley,
S.C.Almo,
and
F.M.Raushel
(2009).
Functional identification of incorrectly annotated prolidases from the amidohydrolase superfamily of enzymes.
|
| |
Biochemistry,
48,
3730-3742.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
D.J.Huggins,
M.D.Altman,
and
B.Tidor
(2009).
Evaluation of an inverse molecular design algorithm in a model binding site.
|
| |
Proteins,
75,
168-186.
|
|
|
|
|
|
|
J.A.Cummings,
A.A.Fedorov,
C.Xu,
S.Brown,
E.Fedorov,
P.C.Babbitt,
S.C.Almo,
and
F.M.Raushel
(2009).
Annotating enzymes of uncertain function: the deacylation of D-amino acids by members of the amidohydrolase superfamily.
|
| |
Biochemistry,
48,
6469-6481.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
J.F.Rakus,
C.Kalyanaraman,
A.A.Fedorov,
E.V.Fedorov,
F.P.Mills-Groninger,
R.Toro,
J.Bonanno,
K.Bain,
J.M.Sauder,
S.K.Burley,
S.C.Almo,
M.P.Jacobson,
and
J.A.Gerlt
(2009).
Computation-facilitated assignment of the function in the enolase superfamily: a regiochemically distinct galactarate dehydratase from Oceanobacillus iheyensis .
|
| |
Biochemistry,
48,
11546-11558.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
K.O.Wrzeszczynski,
and
B.Rost
(2009).
Cell cycle kinases predicted from conserved biophysical properties.
|
| |
Proteins,
74,
655-668.
|
|
|
|
|
|
|
L.Kelly,
U.Pieper,
N.Eswar,
F.A.Hays,
M.Li,
Z.Roe-Zurz,
D.L.Kroetz,
K.M.Giacomini,
R.M.Stroud,
and
A.Sali
(2009).
A survey of integral alpha-helical membrane proteins.
|
| |
J Struct Funct Genomics,
10,
269-280.
|
|
|
|
|
|
|
P.B.Juhl,
P.Trodler,
S.Tyagi,
and
J.Pleiss
(2009).
Modelling substrate specificity and enantioselectivity for lipases and esterases by substrate-imprinted docking.
|
| |
BMC Struct Biol,
9,
39.
|
|
|
|
|
|
|
P.Kolb,
R.S.Ferreira,
J.J.Irwin,
and
B.K.Shoichet
(2009).
Docking and chemoinformatic screens for new ligands and targets.
|
| |
Curr Opin Biotechnol,
20,
429-436.
|
|
|
|
|
|
|
P.Limphong,
M.W.Crowder,
B.Bennett,
and
C.A.Makaroff
(2009).
Arabidopsis thaliana GLX2-1 contains a dinuclear metal binding site, but is not a glyoxalase 2.
|
| |
Biochem J,
417,
323-330.
|
|
|
|
|
|
|
S.D.Copley
(2009).
Prediction of function in protein superfamilies.
|
| |
F1000 Biol Rep,
1,
0.
|
|
|
|
|
|
|
T.C.Terwilliger,
D.Stuart,
and
S.Yokoyama
(2009).
Lessons from structural genomics.
|
| |
Annu Rev Biophys,
38,
371-383.
|
|
|
|
|
|
|
T.Schwede,
A.Sali,
B.Honig,
M.Levitt,
H.M.Berman,
D.Jones,
S.E.Brenner,
S.K.Burley,
R.Das,
N.V.Dokholyan,
R.L.Dunbrack,
K.Fidelis,
A.Fiser,
A.Godzik,
Y.J.Huang,
C.Humblet,
M.P.Jacobson,
A.Joachimiak,
S.R.Krystek,
T.Kortemme,
A.Kryshtafovych,
G.T.Montelione,
J.Moult,
D.Murray,
R.Sanchez,
T.R.Sosnick,
D.M.Standley,
T.Stouch,
S.Vajda,
M.Vasquez,
J.D.Westbrook,
and
I.A.Wilson
(2009).
Outcome of a workshop on applications of protein models in biomedical research.
|
| |
Structure,
17,
151-159.
|
|
|
|
|
|
|
U.Pieper,
N.Eswar,
B.M.Webb,
D.Eramian,
L.Kelly,
D.T.Barkan,
H.Carter,
P.Mankoo,
R.Karchin,
M.A.Marti-Renom,
F.P.Davis,
and
A.Sali
(2009).
MODBASE, a database of annotated comparative protein structure models and associated resources.
|
| |
Nucleic Acids Res,
37,
D347-D354.
|
|
|
|
|
|
|
U.Pieper,
R.Chiang,
J.J.Seffernick,
S.D.Brown,
M.E.Glasner,
L.Kelly,
N.Eswar,
J.M.Sauder,
J.B.Bonanno,
S.Swaminathan,
S.K.Burley,
X.Zheng,
M.R.Chance,
S.C.Almo,
J.A.Gerlt,
F.M.Raushel,
M.P.Jacobson,
P.C.Babbitt,
and
A.Sali
(2009).
Target selection and annotation for the structural genomics of the amidohydrolase and enolase superfamilies.
|
| |
J Struct Funct Genomics,
10,
107-125.
|
|
|
|
|
|
|
C.Kalyanaraman,
H.J.Imker,
A.A.Fedorov,
E.V.Fedorov,
M.E.Glasner,
P.C.Babbitt,
S.C.Almo,
J.A.Gerlt,
and
M.P.Jacobson
(2008).
Discovery of a dipeptide epimerase enzymatic function guided by homology modeling and virtual screening.
|
| |
Structure,
16,
1668-1677.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
D.Dunaway-Mariano
(2008).
Enzyme function discovery.
|
| |
Structure,
16,
1599-1600.
|
|
|
|
|
|
|
H.J.Imker,
J.Singh,
B.P.Warlick,
F.R.Tabita,
and
J.A.Gerlt
(2008).
Mechanistic diversity in the RuBisCO superfamily: a novel isomerization reaction catalyzed by the RuBisCO-like protein from Rhodospirillum rubrum.
|
| |
Biochemistry,
47,
11171-11173.
|
|
|
|
|
|
|
J.Bajorath
(2008).
Computational analysis of ligand relationships within target families.
|
| |
Curr Opin Chem Biol,
12,
352-358.
|
|
|
|
|
|
|
M.Brylinski,
and
J.Skolnick
(2008).
Q-Dock: Low-resolution flexible ligand docking with pocket-specific threading restraints.
|
| |
J Comput Chem,
29,
1574-1588.
|
|
|
|
|
|
|
R.A.Chiang,
A.Sali,
and
P.C.Babbitt
(2008).
Evolutionarily conserved substrate substructures for automated annotation of enzyme superfamilies.
|
| |
PLoS Comput Biol,
4,
e1000142.
|
|
|
|
|
|
|
S.K.Burley,
A.Joachimiak,
G.T.Montelione,
and
I.A.Wilson
(2008).
Contributions to the NIH-NIGMS Protein Structure Initiative from the PSI Production Centers.
|
| |
Structure,
16,
5.
|
|
|
|
|
|
|
W.Tong,
R.J.Williams,
Y.Wei,
L.F.Murga,
J.Ko,
and
M.J.Ondrechen
(2008).
Enhanced performance in prediction of protein active sites with THEMATICS and support vector machines.
|
| |
Protein Sci,
17,
333-341.
|
|
|
|
|
|
|
J.Stubbe
(2007).
Computational biochemistry: models of transition.
|
| |
Nature,
448,
762-763.
|
|
|
|
|
|
|
N.Hertkorn,
C.Ruecker,
M.Meringer,
R.Gugisch,
M.Frommberger,
E.M.Perdue,
M.Witt,
and
P.Schmitt-Kopplin
(2007).
High-precision frequency measurements: indispensable tools at the core of the molecular-level analysis of complex systems.
|
| |
Anal Bioanal Chem,
389,
1311-1327.
|
|
|
|
|
|
|
W.A.Hendrickson
(2007).
Impact of structures from the protein structure initiative.
|
| |
Structure,
15,
1528-1529.
|
|
|
|
|
|
The most recent references are shown first.
Citation data come partly from CiteXplore and partly
from an automated harvesting procedure. Note that this is likely to be
only a partial list as not all journals are covered by
either method. However, we are continually building up the citation data
so more and more references will be included with time.
Where a reference describes a PDB structure, the PDB
codes are
shown on the right.
|
|



 Links
Links












