 |
PDBsum entry 2nvd

|
|
|
|
|
|

|

|

|

|
|

|
|

|
|

|
|

|
|

|
|

|
|
|
 |

|

|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Oxidoreductase
|
PDB id
|
|
|
|
2nvd
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

|

|

|
|
|
|
|
|
|
|
 |
Contents |
 |
|
|
|
|
|
|
|
|
|
|
|
|
* Residue conservation analysis
|
|
|
|
|
|
|
|
|
|
|
Enzyme class 1:
|
|
E.C.1.1.1.21
- aldose reductase.
|
|
|
|
|
|
|
Reaction:
|
|
|
1.
|
an alditol + NAD+ = an aldose + NADH + H+
|
|
2.
|
an alditol + NADP+ = an aldose + NADPH + H+
|
|
|
|
|
|
|
 alditol
alditol
|
+
|
NAD(+)
Bound ligand (Het Group name = )
matches with 91.67% similarity
|
=
|
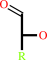 aldose
aldose
|
+
|
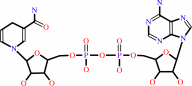 NADH
NADH
|
+
|
H(+)
|
|
|
|
|
|
|
alditol
|
+
|
NADP(+)
Bound ligand (Het Group name = )
corresponds exactly
|
=
|
aldose
|
+
|
 NADPH
NADPH
|
+
|
H(+)
|
|
|
|
|
|
|
|
|
|
Enzyme class 2:
|
|
E.C.1.1.1.300
- NADP-retinol dehydrogenase.
|
|
|
|
|
|
|
Reaction:
|
|
all-trans-retinol + NADP+ = all-trans-retinal + NADPH + H+
|
|
|
|
|
|
all-trans-retinol
Bound ligand (Het Group name = )
matches with 41.94% similarity
|
+
|
NADP(+)
Bound ligand (Het Group name = )
corresponds exactly
|
=
|
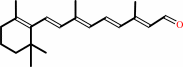 all-trans-retinal
all-trans-retinal
|
+
|
NADPH
|
+
|
H(+)
|
|
|
|
|
|
|
|
|
|
Enzyme class 3:
|
|
E.C.1.1.1.372
- D/L-glyceraldehyde reductase.
|
|
|
|
|
|
|
Reaction:
|
|
|
1.
|
glycerol + NADP+ = L-glyceraldehyde + NADPH + H+
|
|
2.
|
glycerol + NADP+ = D-glyceraldehyde + NADPH + H+
|
|
|
|
|
|
|
 glycerol
glycerol
|
+
|
NADP(+)
Bound ligand (Het Group name = )
corresponds exactly
|
=
|
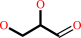 L-glyceraldehyde
L-glyceraldehyde
|
+
|
NADPH
|
+
|
H(+)
|
|
|
|
|
|
|
glycerol
|
+
|
NADP(+)
Bound ligand (Het Group name = )
corresponds exactly
|
=
|
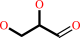 D-glyceraldehyde
D-glyceraldehyde
|
+
|
NADPH
|
+
|
H(+)
|
|
|
|
|
|
|
|
|
|
Enzyme class 4:
|
|
E.C.1.1.1.54
- allyl-alcohol dehydrogenase.
|
|
|
|
|
|
|
Reaction:
|
|
allyl alcohol + NADP+ = acrolein + NADPH + H+
|
|
|
|
|
|
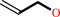 allyl alcohol
allyl alcohol
|
+
|
NADP(+)
Bound ligand (Het Group name = )
corresponds exactly
|
=
|
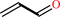 acrolein
acrolein
|
+
|
NADPH
|
+
|
H(+)
|
|
|
|
|
|
|
|
|
|
|
|
|
Note, where more than one E.C. class is given (as above), each may
correspond to a different protein domain or, in the case of polyprotein
precursors, to a different mature protein.
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
DOI no:
|
J Mol Biol
369:186-197
(2007)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
Evidence for a novel binding site conformer of aldose reductase in ligand-bound state.
|
|
H.Steuber,
M.Zentgraf,
C.La Motta,
S.Sartini,
A.Heine,
G.Klebe.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
Human aldose reductase (ALR2) has evolved as a promising therapeutic target for
the treatment of diabetic long-term complications. The binding site of this
enzyme possesses two main subpockets: the catalytic anion-binding site and the
hydrophobic specificity pocket. The latter can be observed in the open or closed
state, depending on the bound ligand. Thus, it exhibits a pronounced capability
for induced-fit adaptations, whereas the catalytic pocket exhibits rigid
properties throughout all known crystal structures. Here, we determined two ALR2
crystal structures at 1.55 and 1.65 A resolution, each complexed with an
inhibitor of the recently described naphtho[1,2-d]isothiazole acetic acid
series. In contrast to the original design hypothesis based on the binding mode
of tolrestat (1), both inhibitors leave the specificity pocket in the closed
state. Unexpectedly, the more potent ligand (2) extends the catalytic pocket by
opening a novel subpocket. Access to this novel subpocket is mainly attributed
to the rotation of an indole moiety of Trp 20 by about 35 degrees . The newly
formed subpocket provides accommodation of the naphthyl portion of the ligand.
The second inhibitor, 3, differs from 2 only by an extended glycolic ester
functionality added to one of its carboxylic groups. However, despite this
slight structural modification, the binding mode of 3 differs dramatically from
that of the first inhibitor, but provokes less pronounced induced-fit
adaptations of the binding cavity. Thus, a novel binding site conformation has
been identified in a region where previous complex structures suggested only low
adaptability of the binding pocket. Furthermore, the two ligand complexes
represent an impressive example of how the slight change of a chemically
extended side-chain at a given ligand scaffold can result in a dramatically
altered binding mode. In addition, our study emphasizes the importance of
crystal structure analysis for the translation of affinity data into
structure-activity relationships.
|
|
|
|
|
|
| |
Selected figure(s)
|
|
|
|
| |
|
|

|
|

|
|
Figure 1.
Figure 1. Chemical formulae of tolrestat (1), two members of
the naphtho[1,2-d]isothiazole acetic acid series (2) and (3),
sorbinil (4), and IDD 594 (5).
|
|
Figure 2.
Figure 2. Three parent binding pocket conformations of ALR2
observed in complexes with sorbinil, tolrestat, and IDD 594
(shown in light blue). Key interactions are shown as red dotted
lines and waters as red spheres. (a) Binding mode of sorbinil
with the specificity pocket in the closed state; the gating
residues Trp 111 and Leu 300 mutually form van der Waals
contacts. (b) In the ALR2–1 complex (tolrestat), Leu 300
adopts a kinked conformation and thereby opens the specificity
pocket. (c) Binding geometry of 5 (IDD 594) with the enzyme;
here, the halogen-substituted aromatic moiety intercalates into
the space created between Trp 111 and Leu 300.
|
|
|
|
|
|
| |
The above figures are
reprinted
by permission from Elsevier:
J Mol Biol
(2007,
369,
186-197)
copyright 2007.
|
|
| |
Figures were
selected
by an automated process.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|
Literature references that cite this PDB file's key reference
|
|
|
| |
PubMed id
|
|
Reference
|
|
|
|
|
|
A.L.Edwards,
and
R.T.Batey
(2009).
A structural basis for the recognition of 2'-deoxyguanosine by the purine riboswitch.
|
| |
J Mol Biol,
385,
938-948.
|
|
|
PDB code:
|
|
|
|
|
|
|
|
J.C.Patra,
and
O.Singh
(2009).
Artificial neural networks-based approach to design ARIs using QSAR for diabetes mellitus.
|
| |
J Comput Chem,
30,
2494-2508.
|
|
|
|
|
|
|
Y.Xu,
J.P.Colletier,
H.Jiang,
I.Silman,
J.L.Sussman,
and
M.Weik
(2008).
Induced-fit or preexisting equilibrium dynamics? Lessons from protein crystallography and MD simulations on acetylcholinesterase and implications for structure-based drug design.
|
| |
Protein Sci,
17,
601-605.
|
|
|
|
|
|
The most recent references are shown first.
Citation data come partly from CiteXplore and partly
from an automated harvesting procedure. Note that this is likely to be
only a partial list as not all journals are covered by
either method. However, we are continually building up the citation data
so more and more references will be included with time.
Where a reference describes a PDB structure, the PDB
code is
shown on the right.
|
|


 Links
Links












