 |
PDBsum entry 2hr1

|
|
|
|
|
|

|

|

|

|
|

|
|

|
|

|
|

|
|

|
|

|
|
|
 |

|

|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Transferase/DNA
|
PDB id
|
|
|
|
2hr1
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

|

|

|
|
|
|
|
|
|
|
 |
Contents |
 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
* Residue conservation analysis
|
|
|
|
|
|
|
|
|
|
|
Enzyme class:
|
|
E.C.2.1.1.37
- Dna (cytosine-5-)-methyltransferase.
|
|
|
|
|
|
|
Reaction:
|
|
a 2'-deoxycytidine in DNA + S-adenosyl-L-methionine = a 5-methyl- 2'-deoxycytidine in DNA + S-adenosyl-L-homocysteine + H+
|
|
|
|
|
|
2'-deoxycytidine in DNA
|
+
|
 S-adenosyl-L-methionine
S-adenosyl-L-methionine
|
=
|
5-methyl- 2'-deoxycytidine in DNA
|
+
|
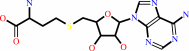 S-adenosyl-L-homocysteine
S-adenosyl-L-homocysteine
|
+
|
H(+)
Bound ligand (Het Group name = )
corresponds exactly
|
|
|
|
|
|
|
|
|
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
DOI no:
|
J Mol Biol
362:516-527
(2006)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
The role of Arg165 towards base flipping, base stabilization and catalysis in M.HhaI.
|
|
F.K.Shieh,
B.Youngblood,
N.O.Reich.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
Arg165 forms part of a previously identified base flipping motif in the
bacterial DNA cytosine methyltransferase, M.HhaI. Replacement of Arg165 with Ala
has no detectable effect on either DNA or AdoMet affinity, yet causes the base
flipping and restacking transitions to be decreased approximately 16 and
190-fold respectively, thus confirming the importance of this motif. However,
these kinetic changes cannot account for the mutant's observed 10(5)-fold
decreased catalytic rate. The mutant enzyme/cognate DNA cocrystal structure
(2.79 A resolution) shows the target cytosine to be positioned approximately 30
degrees into the major groove, which is consistent with a major groove pathway
for nucleotide flipping. The pyrimidine-sugar chi angle is rotated to
approximately +171 degrees, from a range of -95 degrees to -120 degrees in B
DNA, and -77 degrees in the WT M.HhaI complex. Thus, Arg165 is important for
maintaining the cytosine positioned for nucleophilic attack by Cys81. The
cytosine sugar pucker is in the C2'-endo-C3'-exo (South conformation), in
contrast to the previously reported C3'-endo (North conformation) described for
the original 2.70 A resolution cocrystal structure of the WT M.HhaI/DNA complex.
We determined a high resolution structure of the WT M.HhaI/DNA complex (1.96 A)
to better determine the sugar pucker. This new structure is similar to the
original, lower resolution WT M.HhaI complex, but shows that the sugar pucker is
O4'-endo (East conformation), intermediate between the South and North
conformers. In summary, Arg165 plays significant roles in base flipping,
cytosine positioning, and catalysis. Furthermore, the previously proposed
M.HhaI-mediated changes in sugar pucker may not be an important contributor to
the base flipping mechanism. These results provide insights into the base
flipping and catalytic mechanisms for bacterial and eukaryotic DNA
methyltransferases.
|
|
|
|
|
|
| |
Selected figure(s)
|
|
|
|
| |
|
|

|
|

|
|
Figure 1.
Figure 1. (a) Stereo view showing superimposed WT M.HhaI (grey
3MHT.pdb) and R165A M.HhaI (cyan) complexed to AdoHcy (yellow
in WT, orange in R165A), a DNA substrate (red in WT, violet in
R165A) and Val121, Arg165, and Glu119 in green in the WT M.HhaI
complex structure and in marine in the mutant complex
structure. The flipped out cytosine (red in WT, violet in R165A)
can be seen. The RMSD for both structures is 0.47 Å,
based on comparing the protein backbones. (b) The previously
characterized base flipping motif within the WT M.HhaI^26
showing contacts (dotted black lines) between the Arg165 and the
flipped out cytosine base within the active site. DNA
(magenta), and the flipped out cytosine base (salmon); Val121,
Arg165, and Glu119 (green), and the protein backbone (gray) are
shown. The distance unit is Å. (c) Stereo view shows
superimposing the core active sites of the WT M.HhaI/DNA/AdoHcy
(3MHT.pdb grey) and R165A M.HhaI/DNA/AdoHcy (cyan) ternary
structures. The flipped out cytosine base, Cys81, Glu119,
Val121, Arg163, Arg165, and Ala165 (thick lines), and the other
residues and protein backbone (transparent sticks and
transparent cartoon loops) are shown. (d) Stereo view showing
the flipped out cytosine base with the 2F[o]–F[c] map (mesh
in orange) contoured at 1.0 σ in the R165A M.HhaI/DNA/AdoHcy
complex structure. (e) Stereo view showing omit electron
density maps contoured at 1.0 σ, where the flipped out
cytosine base and the 5′ phosphate were omitted in the
structure factor calculation. The heteroatom colors are: red,
oxygen; blue, nitrogen; hot pink, phosphorus; purple, water;
and carbon atoms are shown in gray in all images. Figure 1.
(a) Stereo view showing superimposed WT M.HhaI (grey 3MHT.pdb)
and R165A M.HhaI (cyan) complexed to AdoHcy (yellow in WT,
orange in R165A), a DNA substrate (red in WT, violet in R165A)
and Val121, Arg165, and Glu119 in green in the WT M.HhaI complex
structure and in marine in the mutant complex structure. The
flipped out cytosine (red in WT, violet in R165A) can be seen.
The RMSD for both structures is 0.47 Å, based on comparing
the protein backbones. (b) The previously characterized base
flipping motif within the WT M.HhaI[4]^26 showing contacts
(dotted black lines) between the Arg165 and the flipped out
cytosine base within the active site. DNA (magenta), and the
flipped out cytosine base (salmon); Val121, Arg165, and Glu119
(green), and the protein backbone (gray) are shown. The distance
unit is Å. (c) Stereo view shows superimposing the core
active sites of the WT M.HhaI/DNA/AdoHcy (3MHT.pdb grey) and
R165A M.HhaI/DNA/AdoHcy (cyan) ternary structures. The flipped
out cytosine base, Cys81, Glu119, Val121, Arg163, Arg165, and
Ala165 (thick lines), and the other residues and protein
backbone (transparent sticks and transparent cartoon loops) are
shown. (d) Stereo view showing the flipped out cytosine base
with the 2F[o]–F[c] map (mesh in orange) contoured at 1.0 σ
in the R165A M.HhaI/DNA/AdoHcy complex structure. (e) Stereo
view showing omit electron density maps contoured at 1.0 σ,
where the flipped out cytosine base and the 5′ phosphate were
omitted in the structure factor calculation. The heteroatom
colors are: red, oxygen; blue, nitrogen; hot pink, phosphorus;
purple, water; and carbon atoms are shown in gray in all images.
|
|
Figure 4.
Figure 4. Stereo views of the WT M.HhaI and R165A M.HhaI bound
to cognate DNA, and the WT M.HhaI bound to DNA with constrained
sugar analog substitution.^25 (a)1 The active site of the WT
M.HhaI is shown with Cys81, Glu119, Val121, Arg163, Arg165, and
AdoHcy, which have contacts (dotted red lines) to the flipped
out cytosine base (3MHT.pdb). (a)2 The flipped out cytosine is
positioned in the active site pocket in the WT M.HhaI protein.
(b)1 The active site of the R165A M.HhaI is shown with Cys81,
Glu119, Asn120, Val121, Arg163, Glu164, and Ala165, which have
contacts to the flipped out cytosine base (dotted red lines).
(b)2 The flipped out cytosine base is positioned in the active
site pocket in the R165A M.HhaI protein. (c) The flipped out
South-constrained abasic carbocyclic sugar is rotated in the
active site pocket in the WT M.HhaI protein (1SKM.pdb).The
distance unit is Å and the heteroatom colors are: red,
oxygen; blue, nitrogen; orange, sulfur; hot pink, phosphorus.
Figure 4. Stereo views of the WT M.HhaI and R165A M.HhaI bound
to cognate DNA, and the WT M.HhaI bound to DNA with constrained
sugar analog substitution.[3]^25 (a)1 The active site of the WT
M.HhaI is shown with Cys81, Glu119, Val121, Arg163, Arg165, and
AdoHcy, which have contacts (dotted red lines) to the flipped
out cytosine base (3MHT.pdb). (a)2 The flipped out cytosine is
positioned in the active site pocket in the WT M.HhaI protein.
(b)1 The active site of the R165A M.HhaI is shown with Cys81,
Glu119, Asn120, Val121, Arg163, Glu164, and Ala165, which have
contacts to the flipped out cytosine base (dotted red lines).
(b)2 The flipped out cytosine base is positioned in the active
site pocket in the R165A M.HhaI protein. (c) The flipped out
South-constrained abasic carbocyclic sugar is rotated in the
active site pocket in the WT M.HhaI protein (1SKM.pdb).The
distance unit is Å and the heteroatom colors are: red,
oxygen; blue, nitrogen; orange, sulfur; hot pink, phosphorus.
|
|
|
|
|
|
| |
The above figures are
reprinted
by permission from Elsevier:
J Mol Biol
(2006,
362,
516-527)
copyright 2006.
|
|
| |
Figures were
selected
by an automated process.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|
Literature references that cite this PDB file's key reference
|
|
|
| |
PubMed id
|
|
Reference
|
|
|
|
|
|
F.Xu,
C.Mao,
Y.Ding,
C.Rui,
L.Wu,
A.Shi,
H.Zhang,
L.Zhang,
and
Z.Xu
(2010).
Molecular and enzymatic profiles of mammalian DNA methyltransferases: structures and targets for drugs.
|
| |
Curr Med Chem,
17,
4052-4071.
|
|
|
|
|
|
|
J.A.Castelán-Vega,
A.Jiménez-Alberto,
and
R.M.Ribas-Aparicio
(2010).
Homology modeling and molecular dynamics simulations of HgiDII methyltransferase in complex with DNA and S-adenosyl-methionine: catalytic mechanism and interactions with DNA.
|
| |
J Mol Model,
16,
1213-1222.
|
|
|
|
|
|
|
R.A.Estabrook,
T.T.Nguyen,
N.Fera,
and
N.O.Reich
(2009).
Coupling sequence-specific recognition to DNA modification.
|
| |
J Biol Chem,
284,
22690-22696.
|
|
|
|
|
|
|
H.Demirci,
S.T.Gregory,
A.E.Dahlberg,
and
G.Jogl
(2008).
Crystal structure of the Thermus thermophilus 16 S rRNA methyltransferase RsmC in complex with cofactor and substrate guanosine.
|
| |
J Biol Chem,
283,
26548-26556.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
T.P.Jurkowski,
M.Meusburger,
S.Phalke,
M.Helm,
W.Nellen,
G.Reuter,
and
A.Jeltsch
(2008).
Human DNMT2 methylates tRNA(Asp) molecules using a DNA methyltransferase-like catalytic mechanism.
|
| |
RNA,
14,
1663-1670.
|
|
|
|
|
|
The most recent references are shown first.
Citation data come partly from CiteXplore and partly
from an automated harvesting procedure. Note that this is likely to be
only a partial list as not all journals are covered by
either method. However, we are continually building up the citation data
so more and more references will be included with time.
Where a reference describes a PDB structure, the PDB
codes are
shown on the right.
|
|


 Links
Links













