
|
|
|
|
|
 |
Contents |
 |
|
|
|
|
|
|
|
|
|
|
|
|
* Residue conservation analysis
|
|
|
|
|
|
PDB id:
|
|
|
|
| Name: |
|
Plasmid
|
|
|
Title:
|
|
Ccdb, a topoisomerase poison from e. Coli
|
|
Structure:
|
|
Ccdb. Chain: a, b, c, d. Engineered: yes
|
|
Source:
|
|
Escherichia coli. Organism_taxid: 562. Strain: ms501. Gene: ccdb. Expressed in: escherichia coli. Expression_system_taxid: 562.
|
|
Biol. unit:
|
|
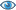 Dimer (from PDB file)
Dimer (from PDB file)
|
|
Resolution:
|
|
|
2.60Å
|
R-factor:
|
0.199
|
R-free:
|
0.246
|
|
|
Authors:
|
|
R.Loris,M.-H.Dao-Thi,E.M.Bahasi,L.Van Melderen,F.Poortmans, R.Liddington,M.Couturier,L.Wyns
|
Key ref:

|
|
R.Loris
et al.
(1999).
Crystal structure of CcdB, a topoisomerase poison from E. coli.
J Mol Biol,
285,
1667-1677.
PubMed id:
DOI:

|
|
|
Date:
|
|
|
17-Apr-98
|
Release date:
|
15-Jul-98
|
|
|
|
|
|
|
PROCHECK
|

|
|
|
|
|
Headers
|
|
|
|
References
|
|
|
|
|
|
|
|
|
|
|
|
Enzyme class:
|
|
Chains A, B, C, D:
E.C.?
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
DOI no:
|
J Mol Biol
285:1667-1677
(1999)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
Crystal structure of CcdB, a topoisomerase poison from E. coli.
|
|
R.Loris,
M.H.Dao-Thi,
E.M.Bahassi,
L.Van Melderen,
F.Poortmans,
R.Liddington,
M.Couturier,
L.Wyns.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
The crystal structure of CcdB, a protein that poisons Escherichia coli gyrase,
was determined in three crystal forms. The protein consists of a five-stranded
antiparallel beta-pleated sheet followed by a C-terminal alpha-helix. In one of
the loops of the sheet, a second small three-stranded antiparallel beta-sheet is
inserted that sticks out of the molecule as a wing. This wing contains the LysC
proteolytic cleavage site that is protected by CcdA and, therefore, forms a
likely CcdA recognition site. A dimer is formed by sheet extension and by
extensive hydrophobic contacts involving three of the five methionine residues
and the C terminus of the alpha-helix. The surface of the dimer on the side of
the alpha-helix is overall negatively charged, while the opposite side as well
as the wing sheet is dominated by positive charges. We propose that the CcdB
dimer binds into the central hole of the 59 kDa N-terminal fragment of GyrA,
after disruption of the head dimer interface of GyrA.
|
|
|
|
|
|
| |
Selected figure(s)
|
|
|
|
| |
|
|

|
|

|
|
Figure 4.
Figure 4. Close up view of the hydrogen bond network around
the buried Asp19 and the adjacent loop Ile25-Arg30. (a) The
consensus structure found in ten out of 13 monomers, with the
peptide bond preceding Pro28 in the cis conformation. Shown is
monomer A of the tetragonal crystal form. (b) The same region in
the monoclinic crystal. (c) The same region in monomers E and G
of the orthorhombic crystal (monomer E is shown). The peptide
bond preceding Pro28 in (b) and (c) is in the trans
conformation. The conformational changes that are observed in
this loop have very little effect on the solvent accessibility
of the buried side-chains of Asp19, Gln21 and Arg31. The
electron density maps correspond with the 2 F[o] -F[c]maps of
the final refined models. A number of side-chains and their
corresponding electron density were omitted for clarity.
|
|
Figure 6.
Figure 6. The interaction between CcdB and GyrA. (a)
Structure of the 59 kDa N-terminal domain of GyrA (GyrA59). The
T-gate domain, which forms the primary dimer interface, is
coloured green, the CAP-like domain red, the tower domain blue,
and the connecting helices grey. Arg462 is shown as a dark grey
CPK model. The position of Gly214 is indicated by a large sphere
around the position of its C^aatom. In this conformation, both
the G-gate and the T-gate are closed and the central hole is not
large enough to accommodate a CcdB dimer in any orientation. (b)
CcdB docked to GyrA59 in the open G-gate conformation, where the
head dimer interface is disrupted. The GyrA59 dimer is shown in
the same colours as in (a), and the CcdB dimer is shown in cyan.
Ample space is present for CcdB binding and CcdB can be rotated
around the 2-fold axis of the complex to increase the contact
area without inducing severe steric clashes.
|
|
|
|
|
|
| |
The above figures are
reprinted
by permission from Elsevier:
J Mol Biol
(1999,
285,
1667-1677)
copyright 1999.
|
|
| |
Figures were
selected
by an automated process.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|
Literature references that cite this PDB file's key reference
|
|
|
| |
PubMed id
|
|
Reference
|
|
|
|
|
|
T.R.Blower,
G.P.Salmond,
and
B.F.Luisi
(2011).
Balancing at survival's edge: the structure and adaptive benefits of prokaryotic toxin-antitoxin partners.
|
| |
Curr Opin Struct Biol,
21,
109-118.
|
|
|
|
|
|
|
T.R.Blower,
X.Y.Pei,
F.L.Short,
P.C.Fineran,
D.P.Humphreys,
B.F.Luisi,
and
G.P.Salmond
(2011).
A processed noncoding RNA regulates an altruistic bacterial antiviral system.
|
| |
Nat Struct Mol Biol,
18,
185-190.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
C.Göbl,
S.Kosol,
T.Stockner,
H.M.Rückert,
and
K.Zangger
(2010).
Solution structure and membrane binding of the toxin fst of the par addiction module.
|
| |
Biochemistry,
49,
6567-6575.
|
|
|
PDB code:
|
|
|
|
|
|
|
|
E.Diago-Navarro,
A.M.Hernandez-Arriaga,
J.López-Villarejo,
A.J.Muñoz-Gómez,
M.B.Kamphuis,
R.Boelens,
M.Lemonnier,
and
R.Díaz-Orejas
(2010).
parD toxin-antitoxin system of plasmid R1--basic contributions, biotechnological applications and relationships with closely-related toxin-antitoxin systems.
|
| |
FEBS J,
277,
3097-3117.
|
|
|
|
|
|
|
J.Yuan,
Y.Sterckx,
L.A.Mitchenall,
A.Maxwell,
R.Loris,
and
M.K.Waldor
(2010).
Vibrio cholerae ParE2 poisons DNA gyrase via a mechanism distinct from other gyrase inhibitors.
|
| |
J Biol Chem,
285,
40397-40408.
|
|
|
|
|
|
|
M.A.Arbing,
S.K.Handelman,
A.P.Kuzin,
G.Verdon,
C.Wang,
M.Su,
F.P.Rothenbacher,
M.Abashidze,
M.Liu,
J.M.Hurley,
R.Xiao,
T.Acton,
M.Inouye,
G.T.Montelione,
N.A.Woychik,
and
J.F.Hunt
(2010).
Crystal structures of Phd-Doc, HigA, and YeeU establish multiple evolutionary links between microbial growth-regulating toxin-antitoxin systems.
|
| |
Structure,
18,
996.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
N.De Jonge,
W.Hohlweg,
A.Garcia-Pino,
M.Respondek,
L.Buts,
S.Haesaerts,
J.Lah,
K.Zangger,
and
R.Loris
(2010).
Structural and thermodynamic characterization of Vibrio fischeri CcdB.
|
| |
J Biol Chem,
285,
5606-5613.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
M.Simic,
N.De Jonge,
R.Loris,
G.Vesnaver,
and
J.Lah
(2009).
Driving forces of gyrase recognition by the addiction toxin CcdB.
|
| |
J Biol Chem,
284,
20002-20010.
|
|
|
|
|
|
|
N.De Jonge,
A.Garcia-Pino,
L.Buts,
S.Haesaerts,
D.Charlier,
K.Zangger,
L.Wyns,
H.De Greve,
and
R.Loris
(2009).
Rejuvenation of CcdB-poisoned gyrase by an intrinsically disordered protein domain.
|
| |
Mol Cell,
35,
154-163.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
A.Garcia-Pino,
M.H.Dao-Thi,
E.Gazit,
R.D.Magnuson,
L.Wyns,
and
R.Loris
(2008).
Crystallization of Doc and the Phd-Doc toxin-antitoxin complex.
|
| |
Acta Crystallogr Sect F Struct Biol Cryst Commun,
64,
1034-1038.
|
|
|
|
|
|
|
K.Bajaj,
M.S.Madhusudhan,
B.V.Adkar,
P.Chakrabarti,
C.Ramakrishnan,
A.Sali,
and
R.Varadarajan
(2007).
Stereochemical Criteria for Prediction of the Effects of Proline Mutations on Protein Stability.
|
| |
PLoS Comput Biol,
3,
e241.
|
|
|
|
|
|
|
M.B.Kamphuis,
M.C.Monti,
R.H.van den Heuvel,
S.Santos-Sierra,
G.E.Folkers,
M.Lemonnier,
R.Díaz-Orejas,
A.J.Heck,
and
R.Boelens
(2007).
Interactions between the toxin Kid of the bacterial parD system and the antitoxins Kis and MazE.
|
| |
Proteins,
67,
219-231.
|
|
|
|
|
|
|
M.Oberer,
K.Zangger,
K.Gruber,
and
W.Keller
(2007).
The solution structure of ParD, the antidote of the ParDE toxin antitoxin module, provides the structural basis for DNA and toxin binding.
|
| |
Protein Sci,
16,
1676-1688.
|
|
|
PDB code:
|
|
|
|
|
|
|
|
N.De Jonge,
L.Buts,
J.Vangelooven,
N.Mine,
L.Van Melderen,
L.Wyns,
and
R.Loris
(2007).
Purification and crystallization of Vibrio fischeri CcdB and its complexes with fragments of gyrase and CcdA.
|
| |
Acta Crystallogr Sect F Struct Biol Cryst Commun,
63,
356-360.
|
|
|
|
|
|
|
A.B.Smith,
and
A.Maxwell
(2006).
A strand-passage conformation of DNA gyrase is required to allow the bacterial toxin, CcdB, to access its binding site.
|
| |
Nucleic Acids Res,
34,
4667-4676.
|
|
|
|
|
|
|
C.Condon
(2006).
Shutdown decay of mRNA.
|
| |
Mol Microbiol,
61,
573-583.
|
|
|
|
|
|
|
M.Inouye
(2006).
The discovery of mRNA interferases: implication in bacterial physiology and application to biotechnology.
|
| |
J Cell Physiol,
209,
670-676.
|
|
|
|
|
|
|
I.Cherny,
L.Rockah,
and
E.Gazit
(2005).
The YoeB toxin is a folded protein that forms a physical complex with the unfolded YefM antitoxin. Implications for a structural-based differential stability of toxin-antitoxin systems.
|
| |
J Biol Chem,
280,
30063-30072.
|
|
|
|
|
|
|
K.Bajaj,
P.Chakrabarti,
and
R.Varadarajan
(2005).
Mutagenesis-based definitions and probes of residue burial in proteins.
|
| |
Proc Natl Acad Sci U S A,
102,
16221-16226.
|
|
|
|
|
|
|
K.Gerdes,
S.K.Christensen,
and
A.Løbner-Olesen
(2005).
Prokaryotic toxin-antitoxin stress response loci.
|
| |
Nat Rev Microbiol,
3,
371-382.
|
|
|
|
|
|
|
K.Kamada,
and
F.Hanaoka
(2005).
Conformational change in the catalytic site of the ribonuclease YoeB toxin by YefM antitoxin.
|
| |
Mol Cell,
19,
497-509.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
Z.Chen,
and
H.Zhao
(2005).
A highly sensitive selection method for directed evolution of homing endonucleases.
|
| |
Nucleic Acids Res,
33,
e154.
|
|
|
|
|
|
|
G.Chakshusmathi,
K.Mondal,
G.S.Lakshmi,
G.Singh,
A.Roy,
R.B.Ch,
S.Madhusudhanan,
and
R.Varadarajan
(2004).
Design of temperature-sensitive mutants solely from amino acid sequence.
|
| |
Proc Natl Acad Sci U S A,
101,
7925-7930.
|
|
|
|
|
|
|
J.A.Smith,
and
R.D.Magnuson
(2004).
Modular organization of the Phd repressor/antitoxin protein.
|
| |
J Bacteriol,
186,
2692-2698.
|
|
|
|
|
|
|
M.H.Dao-Thi,
L.Van Melderen,
E.De Genst,
L.Buts,
A.Ranquin,
L.Wyns,
and
R.Loris
(2004).
Crystallization of CcdB in complex with a GyrA fragment.
|
| |
Acta Crystallogr D Biol Crystallogr,
60,
1132-1134.
|
|
|
|
|
|
|
A.Gogos,
H.Mu,
F.Bahna,
C.A.Gomez,
and
L.Shapiro
(2003).
Crystal structure of YdcE protein from Bacillus subtilis.
|
| |
Proteins,
53,
320-322.
|
|
|
PDB code:
|
|
|
|
|
|
|
|
J.Zhang,
Y.Zhang,
and
M.Inouye
(2003).
Characterization of the interactions within the mazEF addiction module of Escherichia coli.
|
| |
J Biol Chem,
278,
32300-32306.
|
|
|
|
|
|
|
R.Loris,
I.Marianovsky,
J.Lah,
T.Laeremans,
H.Engelberg-Kulka,
G.Glaser,
S.Muyldermans,
and
L.Wyns
(2003).
Crystal structure of the intrinsically flexible addiction antidote MazE.
|
| |
J Biol Chem,
278,
28252-28257.
|
|
|
PDB code:
|
|
|
|
|
|
|
|
D.Hargreaves,
R.Giraldo,
S.Santos-Sierra,
R.Boelens,
D.W.Rice,
R.Díaz Orejas,
and
J.B.Rafferty
(2002).
Crystallization and preliminary X-ray crystallographic studies on the parD-encoded protein Kid from Escherichia coli plasmid R1.
|
| |
Acta Crystallogr D Biol Crystallogr,
58,
355-358.
|
|
|
|
|
|
|
D.Hargreaves,
S.Santos-Sierra,
R.Giraldo,
R.Sabariegos-Jareño,
G.de la Cueva-Méndez,
R.Boelens,
R.Díaz-Orejas,
and
J.B.Rafferty
(2002).
Structural and functional analysis of the kid toxin protein from E. coli plasmid R1.
|
| |
Structure,
10,
1425-1433.
|
|
|
PDB code:
|
|
|
|
|
|
|
|
M.H.Dao-Thi,
D.Charlier,
R.Loris,
D.Maes,
J.Messens,
L.Wyns,
and
J.Backmann
(2002).
Intricate interactions within the ccd plasmid addiction system.
|
| |
J Biol Chem,
277,
3733-3742.
|
|
|
|
|
|
|
K.Gerdes
(2000).
Toxin-antitoxin modules may regulate synthesis of macromolecules during nutritional stress.
|
| |
J Bacteriol,
182,
561-572.
|
|
|
|
|
|
|
D.E.Rawlings
(1999).
Proteic toxin-antitoxin, bacterial plasmid addiction systems and their evolution with special reference to the pas system of pTF-FC2.
|
| |
FEMS Microbiol Lett,
176,
269-277.
|
|
|
|
|
|
|
H.Engelberg-Kulka,
and
G.Glaser
(1999).
Addiction modules and programmed cell death and antideath in bacterial cultures.
|
| |
Annu Rev Microbiol,
53,
43-70.
|
|
|
|
|
|
The most recent references are shown first.
Citation data come partly from CiteXplore and partly
from an automated harvesting procedure. Note that this is likely to be
only a partial list as not all journals are covered by
either method. However, we are continually building up the citation data
so more and more references will be included with time.
Where a reference describes a PDB structure, the PDB
codes are
shown on the right.
|
 |



 Links
Links












