 |
PDBsum entry 1jg0

|
|
|
|
|
 |
Contents |
 |
|
|
|
|
|
|
|
|
|
|
|
|
* Residue conservation analysis
|
|
|
|
|
|
|
|
|
|
|
Enzyme class:
|
|
E.C.2.1.1.45
- thymidylate synthase.
|
|
|
|
|
|
|
Pathway:
|
|
Folate Coenzymes
|
|
|
|
|
|
Reaction:
|
|
dUMP + (6R)-5,10-methylene-5,6,7,8-tetrahydrofolate = 7,8-dihydrofolate + dTMP
|
|
|
|
|
|
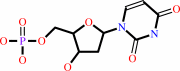 dUMP
dUMP
|
+
|
(6R)-5,10-methylene-5,6,7,8-tetrahydrofolate
Bound ligand (Het Group name = )
corresponds exactly
|
=
|
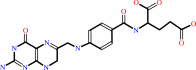 7,8-dihydrofolate
7,8-dihydrofolate
|
+
|
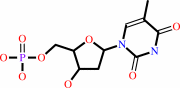 dTMP
dTMP
|
|
|
|
|
|
|
|
|
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
DOI no:
|
Chem Biol
8:981-995
(2001)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
Predicting and harnessing protein flexibility in the design of species-specific inhibitors of thymidylate synthase.
|
|
T.A.Fritz,
D.Tondi,
J.S.Finer-Moore,
M.P.Costi,
R.M.Stroud.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
BACKGROUND: Protein plasticity in response to ligand binding abrogates the
notion of a rigid receptor site. Thus, computational docking alone misses
important prospective drug design leads. Bacterial-specific inhibitors of an
essential enzyme, thymidylate synthase (TS), were developed using a combination
of computer-based screening followed by in-parallel synthetic elaboration and
enzyme assay [Tondi et al. (1999) Chem. Biol. 6, 319-331]. Specificity was
achieved through protein plasticity and despite the very high sequence
conservation of the enzyme between species. RESULTS: The most potent of the
inhibitors synthesized, N,O-didansyl-L-tyrosine (DDT), binds to Lactobacillus
casei TS (LcTS) with 35-fold higher affinity and to Escherichia coli TS (EcTS)
with 24-fold higher affinity than to human TS (hTS). To reveal the molecular
basis for this specificity, we have determined the crystal structure of EcTS
complexed with DDT and 2'-deoxyuridine-5'-monophosphate (dUMP). The 2.0 A
structure shows that DDT binds to EcTS in a conformation not predicted by
molecular docking studies and substantially differently than other TS
inhibitors. Binding of DDT is accompanied by large rearrangements of the protein
both near and distal to the enzyme's active site with movement of C alpha
carbons up to 6 A relative to other ternary complexes. This protein plasticity
results in novel interactions with DDT including the formation of hydrogen bonds
and van der Waals interactions to residues conserved in bacterial TS but not hTS
and which are hypothesized to account for DDT's specificity. The conformation
DDT adopts when bound to EcTS explains the activity of several other LcTS
inhibitors synthesized in-parallel with DDT suggesting that DDT binds to the two
enzymes in similar orientations. CONCLUSIONS: Dramatic protein rearrangements
involving both main and side chain atoms play an important role in the
recognition of DDT by EcTS and highlight the importance of incorporating protein
plasticity in drug design. The crystal structure of the EcTS/dUMP/DDT complex is
a model system to develop more selective TS inhibitors aimed at pathogenic
bacterial species. The crystal structure also suggests a general formula for
identifying regions of TS and other enzymes that may be treated as flexible to
aid in computational methods of drug discovery.
|
|
|
|
|
|
| |
Selected figure(s)
|
|
|
|
| |
|
|

|
|

|
|
Figure 5.
Fig. 5. DDT (N,O-didansyl-L-tyrosine) binding induces dramatic side chain and
backbone plasticity. A: Divergent stereo view showing the
plasticity of the side chains of active site residues between
the EcTS/dUMP/DDT and EcTS/dUMP/ZD1694 structures. DDT and dUMP
are shown in yellow, active site residues from the EcTS/dUMP/DDT
structure are shown in atomic colors and active site residues
from the EcTS/dUMP/ZD1694 structure are shown in gray. B:
Divergent stereo view of aligned monomer backbones of the
EcTS/dUMP/DDT (black and red) and EcTS/dUMP/ZD1694 (green)
structures. The orientation of the structure is the same as that
in A and DDT and dUMP are shown in yellow. Residues in red
mapped onto the EcTS/dUMP/DDT backbone structure are
approximately centrally located residues of protein segments
which shift by > 1Å between the two structures. The
location of the J-helix (J) is also indicated. C: Difference in
Cα position between aligned structures of the EcTS/dUMP/DDT and
EcTS/dUMP/ZD1694 complexes with the position of residues shown
in B indicated.
|
|
Figure 6.
Fig. 6. Ligand-induced, main chain plastic accommodation of
EcTS. Individual panels represent the positional difference
between corresponding Cα carbons of the EcTS/dUMP/mTHF
structure [12] aligned with (A) the EcTS/dUMP/ZD1694 [10], (B)
the EcTS/dUMP/BW1843U89 [11], (C) the EcTS/dUMP/DDT or (D) the
apo EcTS structure [17]. Only one monomer of each of the aligned
structures is shown. D represents the structural changes known
as segmental accommodation that take place upon cofactor binding
to TS.
|
|
|
|
|
|
| |
The above figures are
reprinted
by permission from Cell Press:
Chem Biol
(2001,
8,
981-995)
copyright 2001.
|
|
| |
Figures were
selected
by the author.
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
| |
Protein plasticity in response to ligand binding abrogates the notion of a rigid receptor site. Hence computational docking alone misses important prospective drug design leads. Bacterial-specific inhibitors of an essential enzyme, thymidylate synthase (TS), were developed using a combination of computer-based screening followed by synthetic elaboration. The most potent of the inhibitors synthesized, N,O-didansyl-L-tyrosine (DDT), binds to Lactobacillus casei TS (LcTS) with 35-fold higher affinity and to bacterial TSs with ~30-fold higher affinity than to human TS (hTS). The 2.0Å structure shows that DDT binds to EcTS in a conformation not predicted by molecular docking. Binding of DDT is accompanied by large rearrangements of the protein both near and distal to the enzyme's active site with movement of C alpha carbons up to 6Å relative to other ternary complexes which account for DDT's specificity. Dramatic protein rearrangements involving both main and side chain atoms highlight the importance of incorporating protein plasticity in drug design. The crystal structure also suggests a general formula for identifying regions of proteins that may be treated as flexible to aid in computational methods of drug discovery.
Robert M. Stroud
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
 |
 |
 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Literature references that cite this PDB file's key reference
|
|
|
| |
PubMed id
|
|
Reference
|
|
|
|
|
|
D.D.Robinson,
W.Sherman,
and
R.Farid
(2010).
Understanding kinase selectivity through energetic analysis of binding site waters.
|
| |
ChemMedChem,
5,
618-627.
|
|
|
|
|
|
|
S.Calò,
D.Tondi,
S.Ferrari,
A.Venturelli,
S.Ghelli,
and
M.P.Costi
(2008).
Constrained dansyl derivatives reveal bacterial specificity of highly conserved thymidylate synthases.
|
| |
Chembiochem,
9,
779-790.
|
|
|
|
|
|
|
J.S.Finer-Moore,
A.C.Anderson,
R.H.O'Neil,
M.P.Costi,
S.Ferrari,
J.Krucinski,
and
R.M.Stroud
(2005).
The structure of Cryptococcus neoformans thymidylate synthase suggests strategies for using target dynamics for species-specific inhibition.
|
| |
Acta Crystallogr D Biol Crystallogr,
61,
1320-1334.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
L.Chen,
S.N.Willis,
A.Wei,
B.J.Smith,
J.I.Fletcher,
M.G.Hinds,
P.M.Colman,
C.L.Day,
J.M.Adams,
and
D.C.Huang
(2005).
Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function.
|
| |
Mol Cell,
17,
393-403.
|
|
|
|
|
|
|
K.Gunasekaran,
and
R.Nussinov
(2004).
Modulating functional loop movements: the role of highly conserved residues in the correlated loop motions.
|
| |
Chembiochem,
5,
224-230.
|
|
|
|
|
|
|
A.C.Anderson
(2003).
The process of structure-based drug design.
|
| |
Chem Biol,
10,
787-797.
|
|
|
|
|
|
|
S.J.Teague
(2003).
Implications of protein flexibility for drug discovery.
|
| |
Nat Rev Drug Discov,
2,
527-541.
|
|
|
|
|
|
|
B.K.Shoichet,
S.L.McGovern,
B.Wei,
and
J.J.Irwin
(2002).
Lead discovery using molecular docking.
|
| |
Curr Opin Chem Biol,
6,
439-446.
|
|
|
|
|
|
|
G.M.Verkhivker,
D.Bouzida,
D.K.Gehlhaar,
P.A.Rejto,
S.T.Freer,
and
P.W.Rose
(2002).
Complexity and simplicity of ligand-macromolecule interactions: the energy landscape perspective.
|
| |
Curr Opin Struct Biol,
12,
197-203.
|
|
|
|
|
|
The most recent references are shown first.
Citation data come partly from CiteXplore and partly
from an automated harvesting procedure. Note that this is likely to be
only a partial list as not all journals are covered by
either method. However, we are continually building up the citation data
so more and more references will be included with time.
Where a reference describes a PDB structure, the PDB
codes are
shown on the right.
|
|


 Links
Links











