 |
PDBsum entry 1ss8

|
|
|
|
|
 |
Contents |
 |
|
|
|
|
|
|
|
|
|
|
* Residue conservation analysis
|
|
|
|
|
|
PDB id:
|
|
|
|
| Name: |
|
Chaperone
|
|
|
Title:
|
|
Groel
|
|
Structure:
|
|
Groel protein. Chain: a, b, c, d, e, f, g. Synonym: protein cpn60, 60 kda chaperonin. Engineered: yes
|
|
Source:
|
|
Escherichia coli. Organism_taxid: 562. Gene: grol, groel, mopa, b4143, c5227, z5748, ecs5124, sf4297, s4564. Expressed in: escherichia coli. Expression_system_taxid: 562.
|
|
Biol. unit:
|
|
Heptamer (from PDB file)
|
|
Resolution:
|
|
|
2.70Å
|
R-factor:
|
0.216
|
R-free:
|
0.249
|
|
|
Authors:
|
|
C.Chaudhry,A.L.Horwich,A.T.Brunger,P.D.Adams
|
Key ref:

|
|
C.Chaudhry
et al.
(2004).
Exploring the structural dynamics of the E.coli chaperonin GroEL using translation-libration-screw crystallographic refinement of intermediate states.
J Mol Biol,
342,
229-245.
PubMed id:
DOI:

|
|
|
Date:
|
|
|
23-Mar-04
|
Release date:
|
01-Mar-05
|
|
|
|
|
|
|
PROCHECK
|

|
|
|
|
|
Headers
|
|
|
|
References
|
|
|
|
|
|

|
|
P0A6F5
(CH60_ECOLI) -
Chaperonin GroEL from Escherichia coli (strain K12)
|
|
|
|
Seq:
Struc:
|
|
|
|
548 a.a.
524 a.a.*
|
|

|

|
|
|
|
|
|
|
|
|
|
|
|
Key: |
 |
PfamA domain |
|
|
 |
Secondary structure |
|
 |
CATH domain |
|
|
*
PDB and UniProt seqs differ
at 2 residue positions (black
crosses)
|
|
|
|
|
|
|
|
|
|
|
|
|
Enzyme class:
|
|
E.C.5.6.1.7
- chaperonin ATPase.
|
|
|
|
|
|
|
Reaction:
|
|
ATP + H2O + a folded polypeptide = ADP + phosphate + an unfolded polypeptide
|
|
|
|
|
|
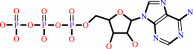 ATP
ATP
|
+
|
H2O
|
+
|
folded polypeptide
|
=
|
 ADP
ADP
|
+
|
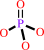 phosphate
phosphate
|
+
|
unfolded polypeptide
|
|
|
|
|
|
|
|
|
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
DOI no:
|
J Mol Biol
342:229-245
(2004)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
Exploring the structural dynamics of the E.coli chaperonin GroEL using translation-libration-screw crystallographic refinement of intermediate states.
|
|
C.Chaudhry,
A.L.Horwich,
A.T.Brunger,
P.D.Adams.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
Large rigid-body domain movements are critical to GroEL-mediated protein
folding, especially apical domain elevation and twist associated with the
formation of a folding chamber upon binding ATP and co-chaperonin GroES. Here,
we have modeled the anisotropic displacements of GroEL domains from various
crystallized states, unliganded GroEL, ATPgammaS-bound, ADP-AlFx/GroES-bound,
and ADP/GroES bound, using translation-libration-screw (TLS) analysis.
Remarkably, the TLS results show that the inherent motions of unliganded GroEL,
a polypeptide-accepting state, are biased along the transition pathway that
leads to the folding-active state. In the ADP-AlFx/GroES-bound folding-active
state the dynamic modes of the apical domains become reoriented and coupled to
the motions of bound GroES. The ADP/GroES complex exhibits these same motions,
but they are increased in magnitude, potentially reflecting the decreased
stability of the complex after nucleotide hydrolysis. Our results have allowed
the visualization of the anisotropic molecular motions that link the static
conformations previously observed by X-ray crystallography. Application of the
same analyses to other macromolecules where rigid body motions occur may give
insight into the large scale dynamics critical for function and thus has the
potential to extend our fundamental understanding of molecular machines.
|
|
|
|
|
|
| |
Selected figure(s)
|
|
|
|
| |
|
|

|
|

|
|
Figure 4.
Figure 4. Effect of intermediate domain motion on apical
domain in unliganded GroEL. For illustrative purposes, we have
performed 25° clockwise and 25° counterclockwise
rotations about the predominant libration axis of the
intermediate domain (I1, see Figure 3). Two orthogonal views
(top and bottom) of the resultant coordinates, related by a
90° vertical rotation are shown to capture the complex
motion. The top views are looking approximately from a
neighboring subunit in the ring, while the bottom views are from
outside the ring looking towards the central 7-fold axis.
|
|
Figure 7.
Figure 7. Intermediate domain motion in the cis GroEL
subunit leads to increased coupling with the equatorial domain.
For illustrative purpose, we have performed 25° clockwise
and 25° counterclockwise rotations about the predominant
libration axis of the intermediate domain (I1, see Figure 6).
|
|
|
|
|
|
| |
The above figures are
reprinted
by permission from Elsevier:
J Mol Biol
(2004,
342,
229-245)
copyright 2004.
|
|
| |
Figures were
selected
by an automated process.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|
Literature references that cite this PDB file's key reference
|
|
|
| |
PubMed id
|
|
Reference
|
|
|
|
|
|
L.Skjaerven,
A.Martinez,
and
N.Reuter
(2011).
Principal component and normal mode analysis of proteins; a quantitative comparison using the GroEL subunit.
|
| |
Proteins,
79,
232-243.
|
|
|
|
|
|
|
F.Zucker,
P.C.Champ,
and
E.A.Merritt
(2010).
Validation of crystallographic models containing TLS or other descriptions of anisotropy.
|
| |
Acta Crystallogr D Biol Crystallogr,
66,
889-900.
|
|
|
|
|
|
|
P.B.Moore
(2009).
On the relationship between diffraction patterns and motions in macromolecular crystals.
|
| |
Structure,
17,
1307-1315.
|
|
|
|
|
|
|
P.D.Kiser,
G.H.Lorimer,
and
K.Palczewski
(2009).
Use of thallium to identify monovalent cation binding sites in GroEL.
|
| |
Acta Crystallogr Sect F Struct Biol Cryst Commun,
65,
967-971.
|
|
|
PDB code:
|
|
|
|
|
|
|
|
R.Potestio,
F.Pontiggia,
and
C.Micheletti
(2009).
Coarse-grained description of protein internal dynamics: an optimal strategy for decomposing proteins in rigid subunits.
|
| |
Biophys J,
96,
4993-5002.
|
|
|
|
|
|
|
K.Hosono,
T.Ueno,
H.Taguchi,
F.Motojima,
T.Zako,
M.Yoshida,
and
T.Funatsu
(2008).
Kinetic Analysis of Conformational Changes of GroEL Based on the Fluorescence of Tyrosine 506.
|
| |
Protein J,
27,
461-468.
|
|
|
|
|
|
|
N.D.Thomsen,
and
J.M.Berger
(2008).
Structural frameworks for considering microbial protein- and nucleic acid-dependent motor ATPases.
|
| |
Mol Microbiol,
69,
1071-1090.
|
|
|
|
|
|
|
A.Korostelev,
and
H.F.Noller
(2007).
Analysis of structural dynamics in the ribosome by TLS crystallographic refinement.
|
| |
J Mol Biol,
373,
1058-1070.
|
|
|
|
|
|
|
A.Korostelev,
S.Trakhanov,
H.Asahara,
M.Laurberg,
L.Lancaster,
and
H.F.Noller
(2007).
Interactions and dynamics of the Shine Dalgarno helix in the 70S ribosome.
|
| |
Proc Natl Acad Sci U S A,
104,
16840-16843.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
J.J.Warren,
T.J.Pohlhaus,
A.Changela,
R.R.Iyer,
P.L.Modrich,
and
L.S.Beese
(2007).
Structure of the human MutSalpha DNA lesion recognition complex.
|
| |
Mol Cell,
26,
579-592.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
P.D.Kiser,
D.T.Lodowski,
and
K.Palczewski
(2007).
Purification, crystallization and structure determination of native GroEL from Escherichia coli lacking bound potassium ions.
|
| |
Acta Crystallogr Sect F Struct Biol Cryst Commun,
63,
457-461.
|
|
|
PDB code:
|
|
|
|
|
|
|
|
S.Mouilleron,
and
B.Golinelli-Pimpaneau
(2007).
Domain motions of glucosamine-6P synthase: comparison of the anisotropic displacements in the crystals and the catalytic hinge-bending rotation.
|
| |
Protein Sci,
16,
485-493.
|
|
|
|
|
|
|
J.Painter,
and
E.A.Merritt
(2006).
Optimal description of a protein structure in terms of multiple groups undergoing TLS motion.
|
| |
Acta Crystallogr D Biol Crystallogr,
62,
439-450.
|
|
|
PDB code:
|
|
|
|
|
|
|
|
K.Suhre,
J.Navaza,
and
Y.H.Sanejouand
(2006).
NORMA: a tool for flexible fitting of high-resolution protein structures into low-resolution electron-microscopy-derived density maps.
|
| |
Acta Crystallogr D Biol Crystallogr,
62,
1098-1100.
|
|
|
|
|
|
|
D.Bourgeois,
and
A.Royant
(2005).
Advances in kinetic protein crystallography.
|
| |
Curr Opin Struct Biol,
15,
538-547.
|
|
|
|
|
|
|
S.Y.Kim,
A.N.Semyonov,
R.J.Twieg,
A.L.Horwich,
J.Frydman,
and
W.E.Moerner
(2005).
Probing the sequence of conformationally induced polarity changes in the molecular chaperonin GroEL with fluorescence spectroscopy.
|
| |
J Phys Chem B,
109,
24517-24525.
|
|
|
|
|
|
The most recent references are shown first.
Citation data come partly from CiteXplore and partly
from an automated harvesting procedure. Note that this is likely to be
only a partial list as not all journals are covered by
either method. However, we are continually building up the citation data
so more and more references will be included with time.
Where a reference describes a PDB structure, the PDB
code is
shown on the right.
|
|


 Links
Links






