 |
PDBsum entry 5c46

|
|
|
|
|
|

|

|

|

|
|

|
|

|
|

|
|

|
|

|
|

|
|

|
|
|
 |

|

|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Transferase/signaling protein
|
PDB id
|
|
|
|
5c46
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|

|

|

|
|
|
|
|
|
|
|
|
|
|
Enzyme class 2:
|
|
Chain E:
E.C.2.7.1.67
- 1-phosphatidylinositol 4-kinase.
|
|
|
|
|
|
|
Pathway:
|
|
1-Phosphatidyl-myo-inositol Metabolism
|
|
|
|
|
|
Reaction:
|
|
a 1,2-diacyl-sn-glycero-3-phospho-(1D-myo-inositol) + ATP = a 1,2-diacyl- sn-glycero-3-phospho-(1D-myo-inositol 4-phosphate) + ADP + H+
|
|
|
|
|
|
1,2-diacyl-sn-glycero-3-phospho-(1D-myo-inositol)
|
+
|
ATP
Bound ligand (Het Group name = )
matches with 90.91% similarity
|
=
|
1,2-diacyl- sn-glycero-3-phospho-(1D-myo-inositol 4-phosphate)
|
+
|
 ADP
ADP
|
+
|
H(+)
|
|
|
|
|
|
|
|
|
|
Enzyme class 3:
|
|
Chain F:
E.C.3.6.5.2
- small monomeric GTPase.
|
|
|
|
|
|
|
Reaction:
|
|
GTP + H2O = GDP + phosphate + H+
|
|
|
|
|
|
GTP
Bound ligand (Het Group name = )
matches with 93.94% similarity
|
+
|
H2O
|
=
|
 GDP
GDP
|
+
|
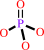 phosphate
phosphate
|
+
|
H(+)
|
|
|
|
|
|
|
|
|
|
|
|
|
Note, where more than one E.C. class is given (as above), each may
correspond to a different protein domain or, in the case of polyprotein
precursors, to a different mature protein.
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
DOI no:
|
Protein Sci
25:826-839
(2016)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
Using hydrogen deuterium exchange mass spectrometry to engineer optimized constructs for crystallization of protein complexes: Case study of PI4KIIIβ with Rab11.
|
|
M.L.Fowler,
J.A.McPhail,
M.L.Jenkins,
G.R.Masson,
F.U.Rutaganira,
K.M.Shokat,
R.L.Williams,
J.E.Burke.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
The ability of proteins to bind and interact with protein partners plays
fundamental roles in many cellular contexts. X-ray crystallography has been a
powerful approach to understand protein-protein interactions; however, a
challenge in the crystallization of proteins and their complexes is the presence
of intrinsically disordered regions. In this article, we describe an application
of hydrogen deuterium exchange mass spectrometry (HDX-MS) to identify dynamic
regions within type III phosphatidylinositol 4 kinase beta (PI4KIIIβ) in
complex with the GTPase Rab11. This information was then used to design
deletions that allowed for the production of diffraction quality crystals.
Importantly, we also used HDX-MS to verify that the new construct was properly
folded, consistent with it being catalytically and functionally active.
Structures of PI4KIIIβ in an Apo state and bound to the potent inhibitor BQR695
in complex with both GTPγS and GDP loaded Rab11 were determined. This hybrid
HDX-MS/crystallographic strategy revealed novel aspects of the PI4KIIIβ-Rab11
complex, as well as the molecular mechanism of potency of a PI4K specific
inhibitor (BQR695). This approach is widely applicable to protein-protein
complexes, and is an excellent strategy to optimize constructs for
high-resolution structural approaches.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|
|



 Links
Links














