 |
PDBsum entry 1op2

|
|
|
|
|
 |
Contents |
 |
|
|
|
|
|
|
|
|
|
|
|
|
* Residue conservation analysis
|
|
|
|
|
References listed in PDB file
|
|
|
Key reference
|
|
|
Title
|
|
Crystal structures and amidolytic activities of two glycosylated snake venom serine proteinases.
|
|
|
Authors
|
|
Z.Zhu,
Z.Liang,
T.Zhang,
Z.Zhu,
W.Xu,
M.Teng,
L.Niu.
|
|
|
Ref.
|
|
J Biol Chem, 2005,
280,
10524-10529.
[DOI no: ]
|
|
|
PubMed id
|
|
|
|
|
|
Abstract
|
|
|
We deduced that Agkistrodon actus venom serine proteinases I and II, previously
isolated from the venom of A. acutus (Zhu, Z., Gong, P., Teng, M., and Niu, L.
(2003) Acta Crystallogr. Sect. D Biol. Crystallogr. 59, 547-550), are encoded by
two almost identical genes, with only the single substitution Asp for Asn at
residue 62. Amidolytic assays indicated that they possess slightly different
enzymatic properties. Crystal structures of A. actus venom serine proteinases I
and II were determined at resolution of 2.0 and 2.1 A with the identification of
trisaccharide (NAG(301)-FUC(302)-NAG(303)) and monosaccharide (NAG(301))
residues in them, respectively. The substrate binding sites S3 of the two
proteinases appear much shallower than that of Trimeresurus stejnegeri venom
plasminogen activator despite the overall structural similarity. Based on
structural analysis, we showed that these Asn(35)-linked oligosaccharides
collide spatially with some inhibitors, such as soybean trypsin inhibitor, and
would therefore hinder their inhibitory binding. Difference of the carbohydrates
in both the proteinases might also lead to their altered catalytic activities.
|
|
|
|

|
|

|
|
Figure 3.
FIG. 3. The saccharides of AaV-SP-I and -II. A and B, the
electron density (2Fo-Fc) maps of NAG301-FUC^302-NAG303 and
NAG301 of AaV-SP-I and AaV-SP-II are contoured at the 1.0  level.
C, superimposed C level.
C, superimposed C  atoms of segments
formed by residues 30-70 for comparing the saccharide residues
between AaV-SP-I (cyan) and -II (yellow). The His57 and the
Asp59 of AaV-SP-I as well as saccharides of the two AaV-SPs are
shown with a stick model. The two hydrogen bonds in AaV-SP-I,
one between FUC^302 and His57 and the other between FUC^302 and
Asp59, are labeled. atoms of segments
formed by residues 30-70 for comparing the saccharide residues
between AaV-SP-I (cyan) and -II (yellow). The His57 and the
Asp59 of AaV-SP-I as well as saccharides of the two AaV-SPs are
shown with a stick model. The two hydrogen bonds in AaV-SP-I,
one between FUC^302 and His57 and the other between FUC^302 and
Asp59, are labeled.
|
|
Figure 5.
FIG. 5. Comparison of the 174 loop and 99 loop in AaV-SP-I
(cyan) AaV-SP-II (yellow), and TSV-PA (silver-gray) shows the
architectural features of substrate binding sites S2  S4 of the
three proteins. The key residues (215, 178, 173, 174, 97, 98,
99) for substrate-binding are shown with stick model. The
hydrogen bonds between Tyr215 and Glu173 of the two AaV-SPs and
the hydrogen bond between Glu97 and Tyr178 of AaV-SP-I are
shown. The amino acid residues of AaV-SPs and TSV-PA are
displayed in black and blue. The schematic substrate-binding
sites 2, 3, and 4 are indicated with a light green ellipse. S4 of the
three proteins. The key residues (215, 178, 173, 174, 97, 98,
99) for substrate-binding are shown with stick model. The
hydrogen bonds between Tyr215 and Glu173 of the two AaV-SPs and
the hydrogen bond between Glu97 and Tyr178 of AaV-SP-I are
shown. The amino acid residues of AaV-SPs and TSV-PA are
displayed in black and blue. The schematic substrate-binding
sites 2, 3, and 4 are indicated with a light green ellipse.
|
|
|
|
|
|
The above figures are
reprinted
by permission from the ASBMB:
J Biol Chem
(2005,
280,
10524-10529)
copyright 2005.
|
|
|
Secondary reference #1
|
|
|
Title
|
|
Purification, N-Terminal sequencing, Partial characterization, Crystallization and preliminary crystallographic analysis of two glycosylated serine proteinases from agkistrodon acutus venom.
|
|
|
Authors
|
|
Z.Zhu,
P.Gong,
M.Teng,
L.Niu.
|
|
|
Ref.
|
|
Acta Crystallogr D Biol Crystallogr, 2003,
59,
547-550.
[DOI no: ]
|
|
|
PubMed id
|
|
|
|
|
|
|
|
|
|

|
|
Figure 1.
Figure 1 Purification of AaV-SP-I and AaV-SP-II from A. acutus
venom. (a) 1.5 g of crude venom was dissolved in 50 ml buffer
A (20 mM Tris-HCl pH 8.2) and left for 2 h at 277 K. The
insoluble materials were removed by centrifugation (4500g) at
277 K for 20 min. The supernatant was then applied to a
DEAE-Sepharose column (1.6 × 40 cm) pre-equilibrated with
buffer A. The effluent was monitored at 280 nm and adjusted to
a flow rate of 134 ml h-1. After the first peak was washed
out, the column was sequentially eluted with a linear gradient
made by mixing 400 ml of buffer A with an equal volume of
buffer B (20 mM Tris-HCl pH 8.2 containing 0.25 M NaCl) and
0.5 M NaCl solution. The effluent was collected for 3 min per
glass tube and investigated for arginine esterase activity.
Three major peaks with arginine esterase activity were observed;
the first peak with arginine esterase activity was pooled,
ultra-filtered and desalted to a volume of 20 ml. The
absorbance at 280 nm is indicated by a solid line, the salt
gradient by a dashed line, arginine esterase activity by squares
and the collected fractions by a solid bar. (b) The protein
fraction pooled from previous chromatography was loaded onto a
CM-Sepharose column (1.8 × 20 cm) pre-equilibrated with a
solution containing 50 mM sodium acetate buffered at pH 5.0 and
then eluted with same solution at a flow rate of 94 ml h-1.
The first two elution peaks (indicated by a solid bar)
possessing arginine esterase activity were collected,
ultra-filtered and desalted to a volume of 10 ml for further
purification. The other materials bound on the column were
eluted with the solution of 0.4 M NaCl (shown by an arrow). (c)
The protein fraction pooled from the second step was applied to
another DEAE-Sepharose column (1.0 × 20 cm) pre-equilibrated
with buffer A. Using a flow rate of 74 ml h-1, the column was
eluted with a linear gradient made by mixing 100 ml of buffer A
with an equal volume of buffer C (20 mM Tris-HCl pH 8.2
containing 0.10 M NaCl). Two protein peaks were found to
possess arginine esterase activity (indicated by solid bars);
the major (on the right) and minor (on the left) components were
designated AaV-SP-I and AaV-SP-II, respectively. Inset in (c):
SDS-PAGE of AaV-SP-I (lanes 1 and 2) and AaV-SP-II (lanes 4 and
5). Lanes 1 and 5 are under non-reducing conditions. Lanes 2 and
4 are under reducing conditions containing 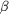 -mercaptoethanol.
Lane 3: markers for molecular-weight estimation. -mercaptoethanol.
Lane 3: markers for molecular-weight estimation.
|
|
|
|
|
|
The above figure is
reproduced from the cited reference
with permission from the IUCr
|
|
|
|
|
|
|




