 |
PDBsum entry 2zmd

|
|
|
|
|
 |
Contents |
 |
|
|
|
|
|
|
|
|
|
|
|
|
* Residue conservation analysis
|
|
|
|
|
|
PDB id:
|
|
|
|
| Name: |
|
Transferase
|
|
|
Title:
|
|
Crystal structure of human mps1 catalytic domain t686a mutant in complex with sp600125 inhibitor
|
|
Structure:
|
|
Dual specificity protein kinase ttk. Chain: a. Fragment: unp residues 510-857, catalytic domain. Synonym: monopolar spindle 1, phosphotyrosine picked threonine- protein kinase, pyt. Engineered: yes. Mutation: yes
|
|
Source:
|
|
Homo sapiens. Human. Organism_taxid: 9606. Gene: ttk, mps1l1. Expressed in: escherichia coli. Expression_system_taxid: 562.
|
|
Resolution:
|
|
|
2.88Å
|
R-factor:
|
0.223
|
R-free:
|
0.261
|
|
|
Authors:
|
|
M.L.H.Chu,L.M.G.Chavas,K.T.Douglas,P.A.Eyers,L.Tabernero
|
Key ref:

|
|
M.L.Chu
et al.
(2008).
Crystal Structure of the Catalytic Domain of the Mitotic Checkpoint Kinase Mps1 in Complex with SP600125.
J Biol Chem,
283,
21495-21500.
PubMed id:
DOI:

|
|
|
Date:
|
|
|
16-Apr-08
|
Release date:
|
13-May-08
|
|
|
|
|
|
|
PROCHECK
|

|
|
|
|
|
Headers
|
|
|
|
References
|
|
|
|
|
|

|
|
P33981
(TTK_HUMAN) -
Dual specificity protein kinase TTK from Homo sapiens
|
|
|
|
Seq:
Struc:
|
|
|
|
857 a.a.
259 a.a.*
|
|

|

|

|
|
|
|
|
|
|
|
|
|
|
Key: |
 |
PfamA domain |
|
|
 |
Secondary structure |
|
 |
CATH domain |
|
|
*
PDB and UniProt seqs differ
at 1 residue position (black
cross)
|
|
|
|
|
|
|
|
|
|
|
|
|
Enzyme class:
|
|
E.C.2.7.12.1
- dual-specificity kinase.
|
|
|
|
|
|
|
Reaction:
|
|
|
1.
|
L-seryl-[protein] + ATP = O-phospho-L-seryl-[protein] + ADP + H+
|
|
2.
|
L-threonyl-[protein] + ATP = O-phospho-L-threonyl-[protein] + ADP + H+
|
|
3.
|
L-tyrosyl-[protein] + ATP = O-phospho-L-tyrosyl-[protein] + ADP + H+
|
|
|
|
|
|
|
L-seryl-[protein]
|
+
|
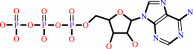 ATP
ATP
|
=
|
O-phospho-L-seryl-[protein]
|
+
|
 ADP
ADP
|
+
|
H(+)
|
|
|
|
|
|
|
L-threonyl-[protein]
|
+
|
ATP
|
=
|
O-phospho-L-threonyl-[protein]
|
+
|
ADP
|
+
|
H(+)
|
|
|
|
|
|
|
L-tyrosyl-[protein]
|
+
|
ATP
|
=
|
O-phospho-L-tyrosyl-[protein]
|
+
|
ADP
|
+
|
H(+)
|
|
|
|
|
|
|
|
|
|
|
|
|
Molecule diagrams generated from .mol files obtained from the
KEGG ftp site
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
|
|
|
|
|
|
| |
|
|
| |
|
DOI no:
|
J Biol Chem
283:21495-21500
(2008)
|
|
PubMed id:
|
|
|
|
|
|
| |
|
Crystal Structure of the Catalytic Domain of the Mitotic Checkpoint Kinase Mps1 in Complex with SP600125.
|
|
M.L.Chu,
L.M.Chavas,
K.T.Douglas,
P.A.Eyers,
L.Tabernero.
|
|
|
|
|
| |
ABSTRACT
|
|
|
|
| |
|
|
Chromosomal instability can result from defective control of checkpoints and is
associated with malignant cell growth. Monopolar spindle 1 (Mps1) is a
dual-specificity protein kinase that has important roles in the prevention of
aneuploidy during the cell cycle and might therefore be a potential target for
new therapeutic agents in the treatment of cancer. To gain insights into the
molecular mechanism of Mps1 inhibition by small molecules, we determined the
x-ray structure of Mps1, both alone and in complex with the ATP-competitive
inhibitor SP600125. Mps1 adopts a classic protein kinase fold, with the
inhibitor sitting in the ATP-binding site where it is stabilized by hydrophobic
interactions. We identified a secondary pocket, not utilized by SP600125, which
might be exploited for the rational design of specific Mps1 inhibitors. These
structures provide important insights into the interaction of this protein
kinase with small molecules and suggest potential mechanisms for Mps1 regulation.
|
|
|
|
|
|
| |
Selected figure(s)
|
|
|
|
| |
|
|

|
|

|
|
Figure 2.
FIGURE 2. Overall structure of human Mps1 catalytic domain.
A, ribbon representation of the structure of Mps1 kinase domain.
Characteristic key features important for substrate binding and
catalysis are labeled as follows: glycine loop (orange),  C helix,
catalytic loop (cyan), and activation loop with DFG motif (blue)
and p + 1 loop (purple). B, comparison of Mps1 apo-WT (green)
and T686A (brown) with phosphorylated Aurora A (P-ArA, red, PDB
ID 1OL5) and unphosphorylated Aurora A (unP-ArA, gray, PDB ID
1MUO). Displacement of helix C, due to the conformation of the
activation loop in the unphosphorylated structures, results in a
shift of the glutamic acid residue and loss of interaction with
the catalytically important lysine. C, superposition of the p +
1 loop between WT apo-kinase (green) and T686A-SP600125 complex
(gold). The positions of Thr-686 and Ala-686 are indicated. C helix,
catalytic loop (cyan), and activation loop with DFG motif (blue)
and p + 1 loop (purple). B, comparison of Mps1 apo-WT (green)
and T686A (brown) with phosphorylated Aurora A (P-ArA, red, PDB
ID 1OL5) and unphosphorylated Aurora A (unP-ArA, gray, PDB ID
1MUO). Displacement of helix C, due to the conformation of the
activation loop in the unphosphorylated structures, results in a
shift of the glutamic acid residue and loss of interaction with
the catalytically important lysine. C, superposition of the p +
1 loop between WT apo-kinase (green) and T686A-SP600125 complex
(gold). The positions of Thr-686 and Ala-686 are indicated.
|
|
Figure 3.
FIGURE 3. Structural insights of the SP600125-binding site.
A, detailed view of the structure of T686A-SP600125 complex,
showing SP600125 bound in the ATP-binding site. The residues
that interact with SP600125 are depicted as sticks. The 2F[o] -
F[c] map around SP600125 is shown, contoured at 1.5  . B,
surface representation (stereo view) of the inhibitor-binding
site in Mps1 and JNK1 (PDB ID 1UKI [PDB]
) with SP600125 shown in yellow. Important differences in the
composition and orientation of key residues
(Mps1^Lys-553/JNK1^Lys-55, Mps1^Cys-604/JNK1^Leu-110,
Mps1^Tyr-591/JNK1^Ile-106, Mps1^Tyr-568, Mps1^Glu-571,
JNK1^Glu-73, JNK1^Met-77, Mps1^Met-602/JNK1^Met-108, and
Mps1^Ile-663/JNK1^Leu-168) can be exploited for the rational
design of Mps1-specific inhibitors. C, surface representation of
the SP600125-binding site in human Mps1. A molecule of PEG from
the crystallization solution is bound in a secondary pocket next
to the catalytic Lys-553. . B,
surface representation (stereo view) of the inhibitor-binding
site in Mps1 and JNK1 (PDB ID 1UKI [PDB]
) with SP600125 shown in yellow. Important differences in the
composition and orientation of key residues
(Mps1^Lys-553/JNK1^Lys-55, Mps1^Cys-604/JNK1^Leu-110,
Mps1^Tyr-591/JNK1^Ile-106, Mps1^Tyr-568, Mps1^Glu-571,
JNK1^Glu-73, JNK1^Met-77, Mps1^Met-602/JNK1^Met-108, and
Mps1^Ile-663/JNK1^Leu-168) can be exploited for the rational
design of Mps1-specific inhibitors. C, surface representation of
the SP600125-binding site in human Mps1. A molecule of PEG from
the crystallization solution is bound in a secondary pocket next
to the catalytic Lys-553.
|
|
|
|
|
|
| |
The above figures are
reprinted
by permission from the ASBMB:
J Biol Chem
(2008,
283,
21495-21500)
copyright 2008.
|
|
| |
Figures were
selected
by an automated process.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
 |
 |
 |
|
Literature references that cite this PDB file's key reference
|
|
|
| |
PubMed id
|
|
Reference
|
|
|
|
|
|
J.Zich,
and
K.G.Hardwick
(2010).
Getting down to the phosphorylated 'nuts and bolts' of spindle checkpoint signalling.
|
| |
Trends Biochem Sci,
35,
18-27.
|
|
|
|
|
|
|
L.Hewitt,
A.Tighe,
S.Santaguida,
A.M.White,
C.D.Jones,
A.Musacchio,
S.Green,
and
S.S.Taylor
(2010).
Sustained Mps1 activity is required in mitosis to recruit O-Mad2 to the Mad1-C-Mad2 core complex.
|
| |
J Cell Biol,
190,
25-34.
|
|
|
|
|
|
|
N.Kwiatkowski,
N.Jelluma,
P.Filippakopoulos,
M.Soundararajan,
M.S.Manak,
M.Kwon,
H.G.Choi,
T.Sim,
Q.L.Deveraux,
S.Rottmann,
D.Pellman,
J.V.Shah,
G.J.Kops,
S.Knapp,
and
N.S.Gray
(2010).
Small-molecule kinase inhibitors provide insight into Mps1 cell cycle function.
|
| |
Nat Chem Biol,
6,
359-368.
|
|
|
PDB codes:
|
|
|
|
|
|
|
|
H.Johnson,
C.E.Eyers,
P.A.Eyers,
R.J.Beynon,
and
S.J.Gaskell
(2009).
Rigorous determination of the stoichiometry of protein phosphorylation using mass spectrometry.
|
| |
J Am Soc Mass Spectrom,
20,
2211-2220.
|
|
|
|
|
|
|
W.Wang,
Y.Yang,
Y.Gao,
Q.Xu,
F.Wang,
S.Zhu,
W.Old,
K.Resing,
N.Ahn,
M.Lei,
and
X.Liu
(2009).
Structural and mechanistic insights into Mps1 kinase activation.
|
| |
J Cell Mol Med,
13,
1679-1694.
|
|
|
PDB code:
|
|
|
|
|
|
|
The most recent references are shown first.
Citation data come partly from CiteXplore and partly
from an automated harvesting procedure. Note that this is likely to be
only a partial list as not all journals are covered by
either method. However, we are continually building up the citation data
so more and more references will be included with time.
Where a reference describes a PDB structure, the PDB
codes are
shown on the right.
|
|


 Links
Links






