Interesting Structures
This tutorial will cover a
spectrum of protein structures available from the Protein
Data Bank and the PDBe
pages. In order to cover this tutorial you will need a graphical
viewer like rasmol installed on your machine. If rasmol is not
available, then java will be required to run java-based viewers such
as AstexViewer
or Jmol. The selection of
structures is not exhaustive and is merely intended to provide a feel
for structures as present in the PDB and their equivalent quaternary
structures in the PQS or PISA
servers. The first link on the legend takes you to the PDBe atlas
pages for this entry, while the second link (View) will start a
java-based viewer.
Example 1

The
crystal structure of the Nucleosome Core Particle (PDB entry: 1AOI)
(View)
The
X-ray crystal structure of the nucleosome core particle of chromatin
shows in atomic detail how the histone protein octamer is assembled
and how 146 base pairs of DNA are organized into a superhelix around
it. Both histone/histone and histone/DNA interactions depend on the
histone fold domains and additional, well ordered structure elements
extending from this motif.
Example
2

The crystal structure of the 20s proteasome from
yeast (PDB entry: 1FNT)(View)

The
crystal structure of archaebacterial 20s proteasome (PDB entry:
1YAR)(View)
The proteasome
from archaebacteria to eukaryotes have the same basic
architecture, which under the electron microscope appears as a
cylinder shaped particle, made up of four stacked rings with
dimensions of approximately 15 nm in height and 11 nm in diameter.
Eukaryotic proteasomes have a more complex structure than the
proteasome from the archaebacterium Thermoplasma acidophilum,
which contains only two different subunits, alpha and beta of 25.8 kD
and 22.3 kD respectively. However, due to gene duplication in the
higher organisms there are 14 different genes which encode 28
subunits that form the eukaryotic proteasome.
Example 3

Crystal
structure of the bovine F1-Atpase (PDB entry: 1BMF)(View)
(Quoted from
http://www.biologie.uni-osnabrueck.de/biophysik/Feniouk/Basics.html)
. Prof.
J.E.Walker received the nobel
prize for chemistry for the determination of the structure of
this enzyme.
During catalysis a complex formed by certain subunits
rotate relative to the rest of the enzyme. This feature makes ATP
synthase the smallest rotary machine ever known.
This enzyme is the primary source of ATP
in a vast majority of living species on Earth, including us. In
human body it daily generates over 100 kg of ATP, which is
subsequently used to provide energy for different biochemical
reactions, including DNA and protein synthesis, muscle contraction,
transport of nutrients and neural activity, to name just a few.
In
plants and photosynthetic bacteria it is essential for solar energy
convertion and carbon fixation. This is one of the oldest enzymes on
Earth, which appeared earlier then photosynthetic or respiratory
enzyme machinery.
This is a membrane
enzyme. It is found in eu- and archebacteria in the plasma membrane;
it is present in the thylacoid membrane in chloroplasts and in the
inner mitochondrial membrane of eucariotic cells. Enzymes from
different organisms show striking homology in the primary structure
of subunits essential for catalysis.
As could be deduced from the name of the enzyme, it
catalyses the reaction of ATP synthesis/hydrolysis. The catalytic act
is coupled with vectoral transmembrane translocation of several
protons. The driving force for ATP synthesis is the transmembrane
electrochemical gradient of protons, while during ATP hydrolysis this
gradient is built using the energy of ATP phosphodietheric bond.
Example 4

Crystal
structure of P53 in complex with DNA (PDB entry: 1TUP)(View)
p53, also known as tumor protein 53
(TP53), is a transcription
factor that regulates the cell cycle and hence functions as a
tumor suppressor. It is very important for cells in multicellular
organisms to suppress cancer. p53 has been described as "the
guardian of the genome" or the "master watchman",
referring to its role in conserving stability by preventing genome
mutation. The name is due to its molecular mass: it runs as a 53
kilodalton (kDa) protein on SDS-PAGE. Mutations in the p53 tumor
suppressor are the most frequently observed genetic alterations in
human cancer. The majority of the mutations occur in the core domain
which contains the sequence-specific DNA binding activity of the p53
protein (residues 102-292), and they result in loss of DNA binding.
p53 has many anti-cancer mechanisms: a) It can activate DNA repair
proteins when DNA has sustained damage. b) It can also hold the cell
cycle at the G1/S regulation point on DNA damage recognition and c)
it can initiate apoptosis, the programmed cell death, if the DNA
damage proves to be irreparable.
Example 4
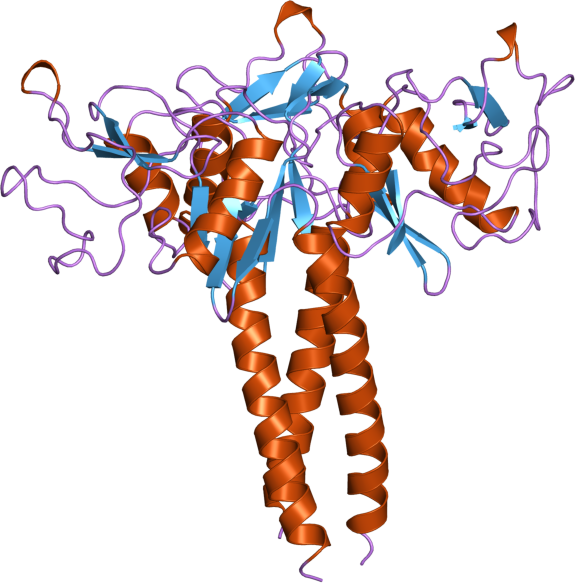
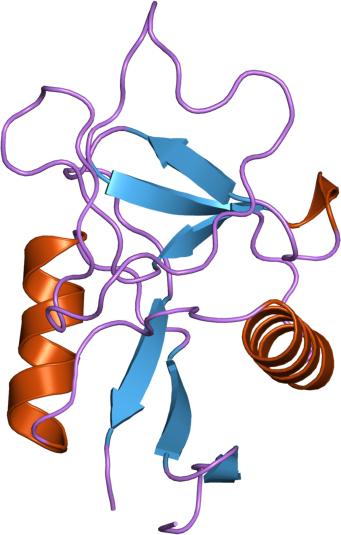
Similar
domains, different functions. On the right (PDB entry: 1LIT),
on the left (PDB entry: 1B08)

Superposed
structure of Lithostathine and Lung Surfactant Protein(SP-D).
Lithostathine is colored in blue and is aligned with one domain of
SP-D
Human Lithostathine (HLIT) (PDB
entry 1LIT) is a pancreatic glycoprotein which inhibits the growth
and nucleation of calcium carbonate crystals. Structural comparison
with the carbohydrate-recognition domains of rat mannose-binding
protein and E-selectin indicates that the C-terminal domain of HLIT
shares a common architecture with the C-type lectins. Nevertheless,
HLIT does not bind carbohydrate nor does it contain the
characteristic calcium-binding sites of the C-type
lectins. In consequence, HLIT represents the first structurally
characterized member of this superfamily which is not a lectin.
Analysis of the charge distribution and calculation of its dipole
moment reveal that HLIT is a strongly polarized molecule. Eight
acidic residues which are separated by regular 6 angstrom spacings
form a unique and continuous patch on the molecular surface. This
arrangement coincides with the distribution of calcium ions on
certain planes of the calcium carbonate crystal; the dipole moment of
HLIT may play a role in orienting the protein on the crystal surface
prior to the more specific interactions of the acidic
residues.
Human lung surfactant protein D (hSP-D) (PDB entry
1B08) belongs to the collectin family of C-type lectins and
participates in the innate immune surveillance against microorganisms
in the lung through recognition of carbohydrate ligands present on
the surface of pathogens. The involvement of this protein in innate
immunity and the allergic response make it the subject of much
interest. The structure comprises an alpha-helical coiled-coil and
three carbohydrate-recognition domains (CRDs) which belong the the
C-type lectin family represented by the the mannan-binding protein
(MBP).
Example 5

The
crystal structure of human placental ribonuclease inhibitor (green)
in comples with human angiogenin (cyan). (PDB entry: 1A4Y)(View)
Human placental RNase inhibitor
(hRI), a leucine-rich repeat protein, binds the blood vessel-inducing
protein human angiogenin (Ang) with extraordinary affinity (Ki >1
fM). The hRI-Ang binding interface is large and encompasses 26
residues from hRI and 24 from Ang, recruited from multiple domains of
both proteins. However, a substantial fraction of the energetically
important contacts involve only a single region of each: the
C-terminal segment 434-460 of hRI and the ribonucleolytic active
centre of Ang, most notably the catalytic residue Lys40.
Example 6
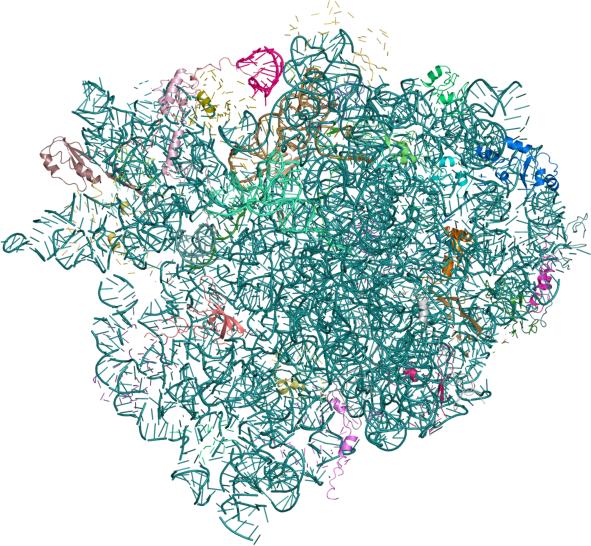
Crystal
structure of the 70s ribosome in complex with mRNA and tRNA (PDB
entries: 2J00
and 2J01)
The
ribosome is the large protein-RNA complex that synthesizes proteins
using genetic instructions encoded in the mRNA template. Ribosomes
are composed of ribosomal
RNA and ribosomal
proteins (known as a Ribonucleoprotein
or RNP). It translates
Messenger RNA (mRNA)
into a polypeptide
chain (e.g., a protein).
It can be thought of as a factory that builds a protein from a set of
genetic instructions. Ribosomes can float freely in the cytoplasm
(the internal fluid of the cell) or bind to the endoplasmic
reticulum, or to the nuclear
envelope. These structures traverse more than one entry due to
the number of protein and nucleic acid chains, as well as the number
of atoms present in the complex. The picture shown above contains 58
chains and over 140000 atoms with coordinates. As a matter of fact,
the experiment determined the structure of two such complexes and is
divided across 4 pdb files (2j00, 2j01, 2j02 and 2j03) and contains
116 chains and over 280000 atoms, thereby making this the largest
structure in the PDB.
Example
7

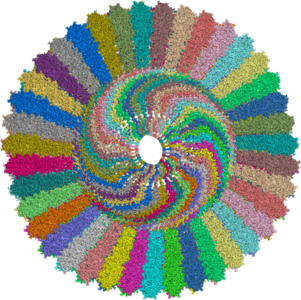
A part of the Rat Liver
Vault determined at 3.50A resolution. (PDB entries: 2ZV4)
From
the Abstract:
Vaults are among the largest cytoplasmic ribonucleoprotein particles
and are found in numerous eukaryotic species. Roles in
multidrug resistance and innate immunity have been
suggested, but the cellular function remains unclear. The
structure of the vault has been determined at 3.5 angstrom resolution
and shows that the cage structure consists of a dimer of
half-vaults, with each half-vault comprising 39 identical
major vault protein (MVP) chains. Each MVP monomer folds
into 12 domains: nine structural repeat domains, a
shoulder domain, a cap-helix domain, and a cap-ring
domain. Interactions between the 42-turn-long cap-helix domains
are key to stabilizing the particle. The shoulder domain is
structurally similar to a core domain of stomatin, a lipid-raft
component in erythrocytes and epithelial cells.
This
protein structure is divided across 3 pdb files (2ZV4, 2ZV5 and
2ZUO). The 3 entries together form half of the vault that is
comprised of 39 identical chains.

The
complete vault can be produced from the crystal structure by applying
a crystallographic symmetry operation on the 39 identical chains in
order to produce a 78 chain complex (as below)