Proctor et al. (2007), Ubiquitin-Proteasome System
December 2008, model of the month by Ranjita Dutta Roy
Original model: BIOMD0000000105
The ubiquitin-proteasome pathway is a major player in the protein degradation system of the body, which is responsible for removing damaged proteins and degrading intact proteins to maintain homeostasis. In short, the purpose of the pathway is to tag proteins with ubiquitin molecules so that they can be recognized by the 26S proteasome, which in turn degrades them. The individual steps of this procedure [1] are outlined in figure 1. The ubiquitin system is involved in various pathogenic mechanisms. For instance, a decline in proteolytic activity has been seen to occur with age. This often leads to accumulation of cross-linked and oxidized proteins and causes protein aggregation, which is the underlying factor of many neurodegenerative diseases such as Alzheimer's and ALS [3].
| The model presented by Proctor et al in the article discussed here [4] is based on a previous model, where the role of the molecular chaperone Hsp90 in aging was investigated [5]. The previous model described how proteins in their correctly folded states can be misfolded with a rate depending on the concentration of reactive oxygen species. Once a protein has been misfolded there are three possible outcomes, 1) it can bind to Hsp90 and either refold and return to the native state or dissociate and remain in the misfolded state, 2) it can be degraded and removed, or 3) it can form an aggregation with another misfolded protein. In the model discussed here, the authors try to determine the primary event in the decline of proteolytic capacity with age by extending the previous model with details of degradation and aggregation. In their present model, Proctor et al take into account the five steps of the ubiquitin-proteasome pathway outlined in figure 1. The interaction between misfolded protein and E3 are considered reversible and the steps involving E1 and E2 are irreversible. These reactions follow stochastic mass action kinetics. They assume that there is a delay between the binding to the proteasome and the degradation. In cases where the protein is not removed or fully degraded, there is a chance that it aggregates with other damaged proteins, which is represented by reactions describing the different ways in which a protein can form aggregates with misfolded proteins bound to ubiquitin and aggregated protein. These aggregates can inhibit the function of the proteasome. Furthermore, the authors included reactions for the rates of protein synthesis, protein misfolding and protein refolding to the model. The rate of protein misfolding depends on the concentration of reactive oxygen species. |  Figure 1: The scheme shows the main steps of the ubiquitin-proteasome pathway. 1) E1 binds to ubiquitin by consumption of one ATP to AMP in order to activate it. 2) The ubiquitin-carrier enzyme E2 takes over the ubiquitin from E1. 3) E2 transfers the ubiquitin to a protein substrate bound to the ubiquitin ligase, E3. 4) The ubiquitin chain is extended. Each step of the extension can be reversed by a deubiquitinating enzyme. 5) A ubiquitinated protein bound with either 1, 2, 3 or 4 ubiquitin molecules binds to the 26 S proteasome in order to be degraded. At degradation of the substrate the ubiquitin molecules are released by deubiquitinating enzymes. Figure taken from [2]. |
The model was validated by mimicking three different conditions: 1) proteasome inhibition, 2) a sudden inactivation of the E1/ubiquitin complex and 3) blocking protein synthesis. The results from the simulations were compared to experimental data of different cell lines. In the in vitro experiments, proteasome inhibition was stimulated by a 26S specific inhibitor and E1 activity was shut off by a temperature sensitive mutation. For each condition, the authors did a sensitivity analysis to see how changes in the parameters affected the steady-state outputs, however, the results will not be discussed in detail here. The model was encoded in SBML and the simulations were done in Biology of Ageing e-Science Integration and Simulation.
 Figure 2: Ubiquitin pools as a function of time under normal conditions. Figure taken from [4]. |  Figure 3: Ubiquitin pools under conditions of proteasome inhibition. Figure taken from [4]. | In general, there was good agreement between the results from time course simulations with experimental data under different conditions. Under normal conditions the native protein pool was fairly stable and the number of misfolded proteins was low. Initially, there was a steep decline of ubiquitin corresponding to the increase of the pools bound to E2 and E1 (see figure 2), and as the system settled down, the pool of ubiquitin conjugates was larger than the pool of monomeric ubiquitin, which agrees with experimental data. The authors also saw that there was no formation of aggregated protein under normal conditions, and as a consequence no inhibition of the proteasome. Under proteasome inhibition the model predicted an initial build-up of ubiquitinated protein. With time the pool of monomeric ubiquitin was depleted, and the level of misfolded proteins increased (see figure 3). This complies with experimental data from IMR20 cells, which show a steady rise in the level of misfolded proteins, and depletion of the ubiquitin pool. However, in ts20 cells there seems to be a compensatory upregulation of ubiquitin. These data suggest that there are differences in the ability to replenish ubiquitin pools between cell lines, and there is a requirement to incorporate ubiquitin turnover rate into the model. The level of native protein is stable, which can be explained by the constant rate of protein synthesis. Furthermore, aggregated proteins are seen to be formed after a few hours giving rise to a low level of aggregates bound to proteasomes. |
By setting the rate of protein synthesis to zero, the authors saw that only the parameters for protein misfolding (green), proteasome activity (red) and shortening the chains of polyubiquitinated misfolded protein bound to the proteasome affect the protein half-life (blue). In figure 4 we see the effect of changing the values of these parameters individually on the decline of total protein. The black line shows the outcome with default values of all parameters.
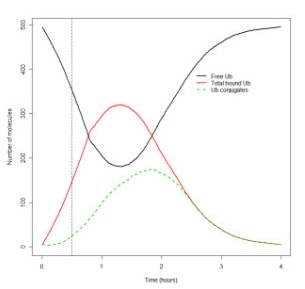 Figure 5: Effects of shutting off E1 activity. Figure taken from [4]. | A temperature sensitive mutation in E1 was used to shut off its activity in ts20 cells (see figure 5). The model predicted that ubiquitin conjugates initially continue being increased after E1 is shut off (dotted line in figure 5), but start to decline after a while and eventually disappear. This followed western blot analysis results, which showed that there was a conversion of conjugated ubiquitin to free ubiquitin after E1 inhibition. |  Figure 4: Effect of individual model parameters on total protein decline. The black line shows the outcome with default values of all parameters. Figure taken from [4]. |
The authors point out that the model should be extended by including details of ubiquitin turnover, models of autophagy, inclusion of a pool of short-lived proteins and adding details of the aggregation process in order to make a predictive model of protein homeostasis and how it is affected in aging. Nevertheless, the present model is a promising initial step as they have managed to show that the model and parameter values are valid under different conditions by comparing to in vitro experiments in several cell lines.
Bibliographic References
- A. Ciechanover. Intracellular Protein Degradation: From a Vague Idea thru the Lysosome and the Ubiquitin-Proteasome System and onto Human Diseases and Drug Targeting. Nobel Lecture, 2004 http://nobelprize.org/nobel_prizes/chemistry/laureates/2004/ciechanover-lecture.html
- Cell Signaling Technology. Ubiquitin/Proteasome Pathway. http://www.cellsignal.com/reference/pathway/Ubiquitin_Proteasome.html, 2007.
- A. Ciechanover. The ubiquitin-proteasome pathway: on protein death and cell life. The EMBO Journal 17(24):7151-7160, 1998. [SRS@EBI]
- C.J. Proctor, M. Tsirigotis, D.A. Gray. An in silico model of the ubiquitin-proteasome system that incorporates normal homeostasis and age-related decline. BMC Syst Biol 1:17, 2007. [SRS@EBI]
- C.J. Proctor, C. Söti, R.J. Boys, C.S. Gillespie, D.P. Shanley, D.J. Wilkinsson, T.B.L. Kirkwood. Modelling the actions of chaperones and their role in ageing. Mech Ageing Dev 126(1):119-131, 2005. [SRS@EBI]